
文章信息
- 李飒, 赵毅强
- LI Sa, ZHAO Yiqiang
- 基因表达数据批次效应去除方法的研究进展
- Research progress on batch effect removal methods for gene expression data
- 南京农业大学学报, 2019, 42(3): 389-397
- Journal of Nanjing Agricultural University, 2019, 42(3): 389-397.
- http://dx.doi.org/10.7685/jnau.201810016
-
文章历史
- 收稿日期: 2018-10-16




高通量技术被广泛应用于生物学研究, 例如鉴定遗传变异体、测定基因和蛋白质表达以及表观遗传修饰[1]。数据的综合分析被认为是从基因组数据集中提取最大有效信息的关键方法[2], 这种方法更有利于研究人员从中发现重要生物学问题。目前, 存在2种不同的综合分析策略:元分析和数据合并。元分析方法首先独立分析每个数据集, 最后将它们的结果联合起来, 获得多个研究结果的综合结论[3]。如果大量研究表明某个结果是重要的, 那么假定这个结果对整体设计研究的问题就是重要的。此外, 在元分析中即使某一结果在某些研究中未达到显著水平, 在其他足够多的研究中达到显著水平, 则最终结果仍然可能是显著的[2, 4]。然而, 对样本量较少的数据集进行元分析时, 所得出的结果容易出现假阳性率高的情况。数据合并的综合分析方法首先将来自不同数据集的样本合并得到一个大的数据集, 再对新合并的数据集进行分析。它比元分析方法的优势主要在于通过分析合并后的大样本集获得的结果具有更高的统计显著性, 推断结果更加严谨, 但是需要开发有效的方法合并来自不同来源的样本集[5]。合并数据的过程中需要考虑不同的时间、平台、实验室等可能造成数据产生差异的条件。
从最早的微阵列试验中就可以观察到批次效应[6-7], 它由许多因素引起, 包括试验分析时间、平台、环境[8]等。当新的样本数据添加到已有的数据集或多个试验数据的元分析中时, 批次效应是不可避免的。为了纠正这些由于非生物条件引起的偏差, 一些研究人员提出了调整数据批次效应的方法。这些调整方法可以是简单的基因水平的均值和方差标准化, 也可以是复杂矩阵分解的方法。如:2000年提出的基于奇异值分解(SVD)的批次数据调整方法, 2004年提出的距离加权判别(DWD)的方法, 2007年提出的替代变量分析(SVA)法和经验贝叶斯方法(即ComBat方法)用于调整批次效应, 2010年提出的基于比值的方法, 2012年提出的两步法(RUV-2)调整批次效应, 2014年基于SVA方法提出了fSVA方法, 2015年基于ComBat方法提出了M-ComBat方法。这些方法可以帮助去除批次效应合并不同批次的数据, 扩大数据量获得更可靠的试验结果。
相同材料不同的技术重复被视为批次, 它们可能来自不同时间、平台、方法、技术和实验室等条件下的测量[9]。由于这些非生物因素的影响容易产生批次效应。很多情况下对不同组的数据进行整合时批次效应容易被忽略。尽管良好的试验设计可以减少批次效应, 但是很难彻底消除[10]。如果没有消除这种系统性偏倚, 当批次效应产生的影响与感兴趣的试验结果相关时, 可能会引起试验结论的偏差, 甚至得出错误的试验结论[11]。因此, 在对数据进行汇集整合时需要选择合适的方法去除批次效应的影响, 然后再进行下游数据分析。
目前, 有很多研究提出了批次效应的去除方法, 不同的方法适用于不同的数据类型, 而基因表达数据是研究最多且应用最为广泛的。本文主要总结介绍已发表文献中的几种去除基因表达数据批次效应的方法及不同方法的适用条件, 并介绍几种对不同批次效应去除方法效果的评估方法。
1 批次效应概念及其潜在来源批次效应表示样品在不同批次中处理和测量产生的与试验期间记录的任何生物变异无关的技术差异[9]。目前, 随着科学问题的深入研究, 需要大量数据用于试验研究。由于不同的平行研究, 或者由于单次试验通量有限而需要分次测量, 导致批次效应的引入。批次效应是高通量试验中常见的变异来源, 受日期、环境、处理组、实验人员、试剂、平台等一些非生物因素的影响, 在试验过程中的每一步都有可能引入批次效应。例如, 测定2个数据集的实验人员不同, 就可能存在实验技术的差异, 进而导致2个数据集之间的差异。实际操作中, 由于一般被记录的只有处理组和日期, 许多可能的差异来源没有被记录, 导致无法明确具体批次效应的来源[1]。如果不妥善处理批次效应可能会严重影响试验结论的准确性。
2 去除批次效应的方法去除基因表达量批次效应的主要方法有ComBat方法、替代变量分析法、距离加权判别法和基于比值的方法等, 下面分述之。
利用合理的统计方法, 可在一定程度上去除批次效应的影响。所有批次效应消除方法都假定批次g的样本j中基因i的测量表达值可以用一般形式表示[9]如下:
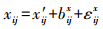
|
(1) |
式中:xij为基因表达量测定值; x′ ij为实际基因表达量; bjx为批次效应值; εijx表示噪声值。
2.1 平均中心方法平均中心方法是通过测量的基因表达值减去样品中每个基因的均值来转化数据[12]。不同批次的基因表达值数据通过中心标准化调整, 使样品中每个基因表达值的平均值变为0。公式如下:
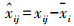
|
(2) |
式中: 
Z-score标准化方法是通过测量的基因表达值减去样品中每个基因的均值再除以样品中每个基因的标准差来转化数据[14-15]。不同批次的基因表达值数据, 通过Z-score标准化方法调整, 使每个基因表达值的均值变为0, 标准差变为1。公式如下:
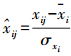
|
(3) |
式中:
基于比值的方法是通过减去每个批次中参考样本的均值来调整不同批次的差异[10, 12]。如果每个批次有多个参考样品, 则使用参考样品的几何平均值或算术平均值为参考[9-10]。公式如下:
基于算术平均值比值方法(Ratio-A):
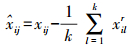
|
(4) |
基于几何平均值比率的方法(Ratio-G):
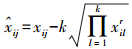
|
(5) |
式中:
距离加权判别(DWD)法是一种对高维低样本量的数据进行两类判别的方法[16-18]。该方法基于支持向量机(SVM)算法, 认为每个批次的样品属于一个特定的分类, 使用DWD作为分类算法, 通过寻找两批次之间的最优超平面w×x+b=0, 分离出不同批次的样品[16]。通过计算每个批次中所有样本到超平面的平均距离(

|
(6) |
式中:
ComBat方法是一种基于经验贝叶斯方法去除批量效应的方法, 尤其对小样本数据更加有效[19-20]。ComBat方法基于估计参数的先验分布, 为每个基因独立估算每个批次的均值和方差并进行调整[21]。测量的基因表达值表现形式如下:

|
(7) |
式中:xijg为批次g(g=1, 2, …, b)样品j(j=1, 2, …, n)基因i(i=1, 2, …, m)的表达测定值; αi为基因i的平均表达值; X为样品条件的设计矩阵; βi为对应X矩阵的回归系数的向量, εijg为误差项假设服从N(0, σi); γig和δig为批次g中基因i加性和乘性批次效应。基因表达数据标准化公式为:

|
(8) |
假设Zijg~N(γig, δig), 利用最小二乘法估计

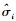
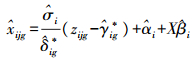
|
(9) |
式中: 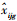
改进的ComBat法(M-ComBat)[14]是将总体样本的平均值和方差转换为“金标准”参考批次的平均值和方差。通过改变整体水平的参数估计值

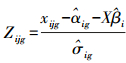
|
(10) |
最后调整数据公式如下:

|
(11) |
式中:g=r为参考批次;
基于奇异值分解(SVD)方法是通过对输入的基因表达值矩阵进行矩阵分解[22-23], 去除与批次效应相关的因子并对矩阵进行重构。几个重要的特征值和相应的特征向量可以捕获大部分的基因表达信息, 通过去除与批次效应相关的特征值来去除批次效应。使用SVD将矩阵因式分解如下:

|
(12) |
式中:Cm×n是Xm×n′和Ym×n″合并的m×n阶矩阵, 其中X和Y表示不同批次样品; m为X和Y样本之间的共同基因的数目; n=n′+n″是X和Y中样本总数; Um×n和(Vn×n)T都是酉矩阵; Um×n的列和(Vn×n)T的行为样品/基因的标准正交基; Σn×n矩阵是包含奇异值的半正定对角矩阵。通过去除相应矩阵中那些被认为与批次效应相对应的分量来完成:

|
(13) |
式中:l≤n; Um×l、Σl×l和(Vn×l)T表示去除批次效应相应分量后的矩阵; 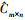
批次效应可能影响几个主要的特征向量, 因此SVD方法不可能识别所有批次效应。并且SVD基于特征向量是正态分布的假设[22], 但引起批次效应来源很多, 并不都服从正态分布, 所以结果会出现不稳定情况。SVD方法不适用于样本量较少的情况。
2.8 替代变量分析(SVA)法替代变量分析(SVA)法直接从表达数据中估计所有未测量因子的影响[24]。SVA算法分为4个基本步骤:1)移除主要变量的贡献获得残差表达矩阵; 2)对残差表达矩阵进行分解(例如SVD或PCA方法)[23, 25-27], 通过残基表达矩阵上的基因与潜在因子之间关联的显著性识别出引起表达变异的基因表达子集, 即与感兴趣的生物因子不相关的其他潜在因子引起的基因表达值变异部分; 3)对每个基因子集, 根据原始表达数据中该子集的批次效应信号构建一个替代变量; 4)重新构建删除批次效应的数据集[24, 28]。通过将sva函数应用于高维数据矩阵来估计替代变量[29]。SVA方法去除批次效应相关因子时, 错误剔除与感兴趣的生物变量相关的因子, 适用于批次分组未知的情况下批次效应的去除。该方法的局限性在于只去除主要的批次效应, 且不适用于样本数目较小的批次校正。
2.9 改进的SVA方法改进的SVA方法(fSVA)[30]首先利用SVA算法校正训练集中的批次效应, 基于训练集应用标准算法建立一个分类器。每次把一个新样品的数据追加到训练集, 使用训练数据集估计的概率权重和系数去除新样品中的批次效应, 利用分类器对去除批次效应的扩增表达矩阵进行分类, 分离出追加的新样品的数据, 得到去除批次效应的新样品表达值。改进SVA方法较SVA方法提高了精度和计算速度。
2.10 两步法两步法(RUV-2)利用1组阴性对照基因来识别与批处理效应相关的因素[31-32]。阴性对照基因是已知与感兴趣生物因子不相关的基因, 一般选取持家基因和spike-in基因, 且持家基因作为阴性对照的表现优于spike-in基因[31]。首先对阴性对照基因进行因子分析, RUV-2方法在这些基因的表达矩阵上应用矩阵分解方法(例如SVD), 通过残基表达矩阵上的基因与潜在因子之间关联的显著性识别出引起表达变异的基因子集, 鉴定与批次效应对应的因子成分; 再利用所得因子调整基因表达值中受潜在变异影响的部分。因为阴性对照基因与感兴趣的因素不相关, 所以不会去除感兴趣的效应。该方法适用于差异表达基因的研究, 通过对阴性对照基因进行因子分析来消除试验过程中产生的无关变化。
3 批量效应去除方法效果的评估非生物因素的影响引起的试验偏差为批次效应, 如不同时间、环境、平台等。如果在整合不同批次的数据时忽略批次效应, 而批次效应产生的影响又与感兴趣的试验结果相关时, 就可能会引起试验结论的偏差, 严重时会影响试验结果的准确性甚至产生错误的试验结果[11]。因此, 在对数据进行汇集整合时需要选择合适的方法去除批次效应的影响, 然后再进行下游数据分析。对批量效应去除方法的效果评估和验证, 和批量效应去除一样重要和困难。如果没有好的可靠的评估工具, 批量效应去除方法可能会引起误差增加甚至导致数据失真, 从而导致任何下游分析的结果出现严重错误[5]。在应用特定去除批次效应方法之前和之后应观察或量化批处理效应的去除效果, 以评估该特定方法是否有效。评估批次效应去除的有效性方法有很多。
3.1 不同批次效应去除方法效果比较Chen等[10]从精确性、准确性和整体性方面对去除批次效应的不同方法进行了评估, 其中包括ComBat方法、SVA方法、基于比值的方法、平均中心方法和DWD方法。首先进行了模拟数据分析, 又通过具体试验数据进行验证。模拟数据集的优势在于已经知道数据集真实的阳性和阴性结果。文献中通过对模拟数据和试验数据进行方差分析, 比较批次效应的校正程度, 评估精确性、准确性和整体性来选择最优方法。综合多种指标最终发现ComBat方法优于其他方法, 批次效应去除效果最好。
3.2 评估批次效应去除效果的方法基因表达数据箱线图[1, 5, 33]通过5个样品参数(极大、小值, 中位数, 上、下四分位数)表示出样本总体分布情况, 根据箱线图的分布情况判断调整方法的有效性。箱线图分布越靠近调整效果越好。批次效应去除前、后基因表达箱线图见图 1。批次效应去除前2个批次数据之间的表达值分布存在差别, 批次效应去除后表达值分布更趋向一致。

|
图 1 批次效应去除前、后不同批次数据基因表达值分布图 Fig. 1 Distribution of gene expression values for different batches of data before and after batch effect removal None表示未经过批次效应调整的数据; ComBat表示经过ComBat方法调整过的数据; Mean center(平均中心)表示经过平均中心方法调整过的数据。数据来自GEO数据库的GSE10072和GSE19804, 基因为MMY14。经ComBat方法和平均中心方法调整后数据分布更整齐。None means data that were not adjusted for the batch effect; ComBat means data that were adjusted for the ComBat method; Mean center means data that were adjusted for the Mean center method. Data are collected from GSE10072 and GSE19804 in the GEO database, and the gene is MMY 14. The data distribution between different samples are more orderly after the ComBat method and Mean center adjusted. |
通过估计不同批次基因表达值的密度分布绘制基因表达分布密度图[33], 根据概率密度函数图的重叠情况判断调整方法的有效性, 密度曲线重叠程度越高表示调整效果越好。批次效应去除前、后密度分布图见图 2。批次效应去除前2个批次的基因表达分布密度图之间存在差别, 经ComBat方法调整后不同批次基因表达密度图基本重叠, 平均中心方法调整后不同批次基因表达密度图位置重叠峰值不同。ComBat方法比平均中心方法调整效果好。

|
图 2 批次效应去除前、后不同批次数据基因表达值密度分布图 Fig. 2 Density distribution map of gene expression values of different batches before and after batch effect removal 经ComBat方法调整后不同批次基因表达密度图基本重叠, 平均中心方法调整后不同批次基因表达密度图位置重叠峰值不同。ComBat方法比平均中心方法调整效果好。After the adjustment of the ComBat method, the expression density maps of different batches of genes basically overlap, after the adjustment of the Mean center method, the positions of different batches of gene expression density maps overlap, and the peaks are different. The ComBat method is better than the Mean center method. |
通过对不同批次数据分层聚类获得树状图[33]。树状图通常用于聚类同类群中的基因或样本, 拥有相同生物学特性的样品应该聚类在一起。该方法从单个样品方面判断批次效应去除效果。如果样品按批次聚类, 表明有批次效应。批次效应去除前、后聚类树形图见图 3。批次效应去除前来自于同一批次的数据分别聚成一类, ComBat方法和平均中心方法调整后, 来自同一批次的数据不聚类在一起, 批次效应不再明显。

|
图 3 批次效应去除前、后不同批次数据聚类树状图 Fig. 3 Clustering tree plots with different batches before and after batch effect removal 数据来自GEO数据库的GSE10072和GSE19804, 其中GSM254722—GSM254731样品来自GSE10072, GSM494556—GSM494565样品来自GSE19804。经ComBat方法和平均中心方法调整后, 来自同一批次的数据不聚类在一起。Data were collected from GSE10072 and GSE19804 of GEO database, among which GSM254716-GSM254731 samples come from GSE10072, and GSM494556-GSM494571 samples come from GSE19804. Data from different samples were not clustered in batches after the ComBat method and Mean center method adjusted. |
对基因表达矩阵进行对数转化, 计算每个基因的对数表达值及其中值, 计算每个样品中每个基因的对数表达值与中值的偏差, 根据偏差值绘制相对对数表达图[34]。根据偏差值的大小从单个样品方面判断调整方法的有效性, 箱线图以0为中心, 其宽度越小表示调整效果越好。批次效应去除前、后相对对数表达图见图 4。批次效应去除前各数据之间偏差较大, 经ComBat方法调整后, 样品箱线图的宽度变小, 批次效应去除效果明显, 平均中心方法调整后, 样品箱线图的宽度变大, 数据分布更不整齐, 对于单个样品批次效应没有有效去除。

|
图 4 批次效应去除前、后相对对数表达图 Fig. 4 Relative log expression before and after the removal of batch effect 数据来自GEO数据库的GSE10072和GSE19804, 其中样品为GSM254716—GSM254731来自GSE10072, 样品GSM494556—GSM494571来自GSE19804。Data were collected from GSE10072 and GSE19804 of GEO database, among which GSM254722-GSM254731 samples come from GSE10072, and GSM494556-GSM494565 samples come from GSE19804. |
可视化方法可以对批次效应去除方法的有效性进行粗略的估计, 并且能对去除结果进行快速检查, 因此这类方法在实际应用中是最常见最直接的。另外, 也可以用定量度量的方法准确估计批次效应去除效果。例如, 主方差成分分析法(PVCA)[35]结合主成分分析(PCA)和方差分量分析(VCA)2种方法, 判断变异来源, 并对其进行量化。根据量化结果判断调整方法是否有效, 调整后代表批次效应的变异来源比率越小调整效果越好。混合分数(mixture score)方法[36-37]通过计算数据集M中每个样品在k-近邻范围内属于数据集N的样品数与M中样品数k的比值。公式如下:

|
(14) |
式中:x是N的样品在M的k-近邻范围内的数, 0≤Mixture score≤1。Mixture score越接近0.5说明2个批次的数据重复度越好, 批次效应去除效果越好, 接近0或1表示数据重复度不好, 批次效应越明显。
4 结论与展望整合来自不同试验的数据毫无疑问是大规模基因组数据分析的有效方法。目前, 有很多文献提出了去除批次效应的方法, 不同方法适用于不同的数据类型。其中, 平均中心和Z-score标准化方法对数据进行了简单的转换, 但在批次效应复杂的情况下不能有效去除批次效应。基于比例的方法通过1个或1组对照或参考样本的平均值来缩放表达值, 应用于预测模型数据调整时优于其他方法。距离加权判别法1次只能分析2个批次, 对于大型研究并不方便。ComBat方法提供清晰的数据模型, 已知批次分组时可以有效去除批次效应, 并且可以有效处理样本量较小的数据批次效应。SVA方法可以有效避免去除批次效应相关因子时移除与感兴趣的生物效应相关的因子。RUV-2方法则适用于差异表达基因分析中去除批次效应。通过试验数据和模拟数据分析, ComBat方法调整效果最好, 其次是平均中心方法。小样本量时, 只有ComBat方法适用。目前, 很多去除批次效应的方法被开发, 并且在确定和计量批次效应方面取得了实质性进展, 但仍然存在巨大的挑战。针对不同的试验, 批次效应的来源和影响可能有很大不同, 需要分析批处理效应最常见的潜在来源, 使之更有效去除批次效应, 整合来自不同批次的数据。同时, 需要更有效的办法准确判断出数据中批次效应影响的部分, 更准确去除批次效应。对批次处理方法进行扩展应用于其他数据, 例如基因组数据, 最终能够有效增加数据量, 充分利用不同试验来源的数据得到更可靠的结果, 以提升大数据分析的能力。
| [1] |
Leek J T, Scharpf R B, Bravo H C, et al. Tackling the widespread and critical impact of batch effects in high-throughput data[J]. Nat Rev Genet, 2010, 11(10): 733-739. DOI:10.1038/nrg2825 |
| [2] |
Rhodes D R, Chinnaiyan A M. Integrative analysis of the cancer transcriptome[J]. Nat Genet, 2005, 37: S31-S37. DOI:10.1038/ng1570 |
| [3] |
Barendregt J J, Doi S A, Lee Y Y, et al. Meta-analysis of prevalence[J]. Journal of Epidemiology and Community Health, 2013, 67(11): 974-978. DOI:10.1136/jech-2013-203104 |
| [4] |
Rhodes D R, Yu J, Shanker K, et al. Large-scale meta-analysis of cancer microarray data identifies common transcriptional profiles of neoplastic transformation and progression[J]. Proc Natl Acad Sci USA, 2004, 101(25): 9309-9314. DOI:10.1073/pnas.0401994101 |
| [5] |
Lazar C, Meganck S, Taminau J, et al. Batch effect removal methods for microarray gene expression data integration:a survey[J]. Brief Bioinform, 2013, 14(4): 469-490. DOI:10.1093/bib/bbs037 |
| [6] |
Lander E S. Array of hope[J]. Nature Genetics, 1999, 21(51): 3-4. |
| [7] |
Akey J M, Biswas S, Leek J T, et al. On the design and analysis of gene expression studies in human populations[J]. Nat Genet, 2007, 39(7): 807-809. DOI:10.1038/ng0707-807 |
| [8] |
Fare T L, Coffey E M, Dai H, et al. Effects of atmospheric ozone on microarray data quality[J]. Anal Chem, 2003, 75(17): 4672-4675. DOI:10.1021/ac034241b |
| [9] |
Hornung R, Boulesteix A L, Causeur D. Combining location-and-scale batch effect adjustment with data cleaning by latent factor adjustment[J]. BMC Bioinformatics, 2016, 17: 27. DOI:10.1186/s12859-015-0870-z |
| [10] |
Chen C, Grennan K, Badner J, et al. Removing batch effects in analysis of expression microarray data:an evaluation of six batch adjustment methods[J]. PLoS One, 2011, 6(2): e17238. DOI:10.1371/journal.pone.0017238 |
| [11] |
Nyamundanda G, Poudel P, Patil Y, et al. A novel statistical method to diagnose, quantify and correct batch effects in genomic studies[J]. Sci Rep, 2017, 7(1): 10849. DOI:10.1038/s41598-017-11110-6 |
| [12] |
Luo J, Schumacher M, Scherer A, et al. A comparison of batch effect removal methods for enhancement of prediction performance using MAQC-Ⅱ microarray gene expression data[J]. Pharmacogenomics J, 2010, 10(4): 278-291. DOI:10.1038/tpj.2010.57 |
| [13] |
Sims A H, Smethurst G J, Hey Y, et al. The removal of multiplicative, systematic bias allows integration of breast cancer gene expression datasets-improving meta-analysis and prediction of prognosis[J]. BMC Med Genomics, 2008, 1: 42. DOI:10.1186/1755-8794-1-42 |
| [14] |
Stein C K, Qu P, Epstein J, et al. Removing batch effects from purified plasma cell gene expression microarrays with modified ComBat[J]. BMC Bioinformatics, 2015, 16: 63. DOI:10.1186/s12859-015-0478-3 |
| [15] |
Curtis A E, Smith T A, Ziganshin B A, et al. The mystery of the Z-score[J]. Aorta, 2016, 4(4): 124-130. DOI:10.12945/j.aorta.2016.16.014 |
| [16] |
Benito M, Parker J, Du Q, et al. Adjustment of systematic microarray data biases[J]. Bioinformatics, 2004, 20(1): 105-114. |
| [17] |
Huang H, Lu X, Liu Y, et al. R/DWD:distance-weighted discrimination for classification, visualization and batch adjustment[J]. Bioinformatics, 2012, 28(8): 1182-1183. DOI:10.1093/bioinformatics/bts096 |
| [18] |
Zhang Y, Ren J, Jiang J. Combining MLC and SVM classifiers for learning based decision making:analysis and evaluations[J]. Comput Intell Neurosci, 2015, 2015: 1-8. |
| [19] |
Johnson W E, Li C, Rabinovic A. Adjusting batch effects in microarray expression data using empirical Bayes methods[J]. Biostatistics, 2007, 8(1): 118-127. DOI:10.1093/biostatistics/kxj037 |
| [20] |
Muller C, Schillert A, Rothemeier C, et al. Removing batch effects from longitudinal gene expression-quantile normalization plus comBat as best approach for microarray transcriptome data[J]. PLoS One, 2016, 11(6): e0156594. DOI:10.1371/journal.pone.0156594 |
| [21] |
Li C, Wong W H. Model-based analysis of oligonucleotide arrays:expression index computation and outlier detection[J]. Proc Natl Acad Sci USA, 2001, 98(1): 31-36. DOI:10.1073/pnas.98.1.31 |
| [22] |
Zhang W, Xiao F, Li B, et al. Using SVD on clusters to improve precision of interdocument similarity measure[J]. Comput Intell Neurosci, 2016, 2016: 1-11. |
| [23] |
Alter O, Brown P O, Botstein D. Singular value decomposition for genome-wide expression data processing and modeling[J]. Proc Natl Acad Sci USA, 2000, 97(18): 10101-10106. DOI:10.1073/pnas.97.18.10101 |
| [24] |
Leek J T, Storey J D. Capturing heterogeneity in gene expression studies by surrogate variable analysis[J]. PLoS Genet, 2007, 3(9): e161. DOI:10.1371/journal.pgen.0030161 |
| [25] |
Jolliffe I T. Principal Component Analysis[M]. 2nd ed. New York: Springer, 2002: 487.
|
| [26] |
Jolliffe I T. Principal Component Analysis[M]. New York: Springer-Verlag, 1986: 271.
|
| [27] |
Reese S E, Archer K J, Therneau T M, et al. A new statistic for identifying batch effects in high-throughput genomic data that uses guided principal component analysis[J]. Bioinformatics, 2013, 29(22): 2877-2883. DOI:10.1093/bioinformatics/btt480 |
| [28] |
Leek J T. Svaseq:removing batch effects and other unwanted noise from sequencing data[J]. Nucleic Acids Res, 2014, 42(21): e161. DOI:10.1093/nar/gku864 |
| [29] |
Leek J T, Johnson W E, Parker H S, et al. The sva package for removing batch effects and other unwanted variation in high-throughput experiments[J]. Bioinformatics, 2012, 28(6): 882-883. DOI:10.1093/bioinformatics/bts034 |
| [30] |
Parker H S, Corrada B H, Leek J T. Removing batch effects for prediction problems with frozen surrogate variable analysis[J]. Peer J, 2014, 2: e561. DOI:10.7717/peerj.561 |
| [31] |
Gagnon-Bartsch J A, Speed T P. Using control genes to correct for unwanted variation in microarray data[J]. Biostatistics, 2012, 13(3): 539-552. DOI:10.1093/biostatistics/kxr034 |
| [32] |
Jacob L, Gagnon-Bartsch J A, Speed T P. Correcting gene expression data when neither the unwanted variation nor the factor of interest are observed[J]. Biostatistics, 2016, 17(1): 16-28. |
| [33] |
Kim K Y, Kim S H, Ki D H, et al. An attempt for combining microarray data sets by adjusting gene expressions[J]. Cancer Res Treat, 2007, 39(2): 74-81. DOI:10.4143/crt.2007.39.2.74 |
| [34] |
Gandolfo L C, Speed T P. RLE plots:visualizing unwanted variation in high dimensional data[J]. PLoS One, 2018, 13(2): e0191629. DOI:10.1371/journal.pone.0191629 |
| [35] |
Boedigheimer M J, Wolfinger R D, Bass M B, et al. Sources of variation in baseline gene expression levels from toxicogenomics study control animals across multiple laboratories[J]. BMC Genomics, 2008, 9: 285. DOI:10.1186/1471-2164-9-285 |
| [36] |
Kim K Y, Kim S H, Ki D H, et al. An attempt for combining microarray data sets by adjusting gene expressions[J]. Cancer Res Treat, 2007, 39(2): 74-81. DOI:10.4143/crt.2007.39.2.74 |
| [37] |
Kim K Y, Ki D H, Jeong H J, et al. Novel and simple transformation algorithm for combining microarray data sets[J]. BMC Bioinformatics, 2007, 8(1): 218. DOI:10.1186/1471-2105-8-218 |