 2011, Vol. 47
2011, Vol. 47文章信息
- 刘恩英, 王源秀, 徐立安, 黄敏仁
- Liu Enying, Wang Yuanxiu, Xu Li'an, Huang Minren
- 基于SSR和SRAP标记的簸箕柳×绵毛柳遗传框架图
- A Genetic Frame Map of Salix suchowensis × S.erioclada Based on SSR and SRAP Markers
- 林业科学, 2011, 47(5): 23-30.
- Scientia Silvae Sinicae, 2011, 47(5): 23-30.
-
文章历史
- 收稿日期:2010-01-28
- 修回日期:2010-03-24
-
作者相关文章



2. 安徽师范大学生命科学学院 芜湖 241000;
3. Forest Biotechnology Group,North Carolina State University Raleigh NC 27695 USA
2. College of Life Sciences, Anhui Normal University Wuhu 241000;
3. Forest Biotechnology Group, North Carolina State University Raleigh NC 27695 USA
柳属(Salix)树种丰富,分布广泛。柳树耐水湿,生长快、生物量高,是我国重要的阔叶树种; 在欧洲和美国,作为短轮伐矮林(short-rotation coppice,SRC)能源树种被广泛栽培。柳树基因组较小(同杨树相似,约500 Mb),易进行有性和无性繁殖,灌木柳1年生即能开花结实,可弥补林木世代周期长的缺陷,是进行基因组和木本植物遗传学研究的理想材料(涂忠虞,1982; 王源秀等,2008)。
自21世纪初始,国外已进行了蒿柳(S.viminalis)、毛枝柳(S.dasyclados)、白柳(S.alba)、爆竹柳(S.fragilis)等树种的遗传图谱构建研究(Tsarouhas et al., 2002; Hanley et al., 2002; 2006; Barcaccia et al., 2003; Rǒnnberg-Wstljung et al., 2003; Berlin et al., 2010)。初期的作图标记以RFLP,AFLP为主,后来SSR等标记得到愈来愈多的应用,其中相当部分SSR标记来自柳属的近缘属杨属(Populus)。杨树是林木基因组学研究的模式树种,具有良好的基因组学和分子生物学研究基础,如高分辨率的参照图谱、全基因组序列、大量通用性较好的分子标记和巨大的EST数据库,为杨柳科基因组比较研究提供了平台(Tuskan et al., 2006)。Hanley等(2006)利用所构建的蒿柳遗传连锁图谱与杨树基因组进行比较,发现柳树与杨树基因组具有高度同源性,且存在较高的同线性(synteny)、共线性(collinearity)及微共线性(microsynteny)。
本研究利用杨树和柳树的通用性SSR分子标记以及SCAR和SRAP标记,对簸箕柳(S.suchowensis)、绵毛柳(S.erioclada)及F1群体进行遗传分析,构建柳树遗传连锁图谱,并与毛果杨(Populus trichocarpa)基因组进行初步比较分析,希望能为柳树高密度遗传图谱构建、QTLs定位和杨柳科内比较基因组的研究提供一定的基础。
1 材料与方法 1.1 引物来源杨树SSR引物1 656对,信息来源于http://wwwornlgov/sci/ipgc/ssr_resourcehtm(USA); 68对柳树SSR引物、2对SCAR标记引物来自NCBI数据库和文献(邓玉营等,2008),SRAP的正反向引物各10条选自文献(杨琦等,2007)。标记采用原始名称。
1.2 作图群体作图群体为簸箕柳295-2×绵毛柳716的F1群体。亲本簸箕柳295-2和绵毛柳716及其198个F1子代由江苏省林业科学研究院潘明建研究员提供,母本簸箕柳295-2是20世纪60年代采于苏州,父本绵毛柳716由美国纽约州Vassar大学的Fritz博士1998年赠送。簸箕柳和绵毛柳均为灌木柳(2n=38),分别来自柳属的筐柳组(Sect.Helix)和绵毛柳组(Sect.Eriocladae),性状差异较大。所有研究材料于2004年栽植于南京林业大学实验苗圃。
2006年4月调查3年生F1实生苗的性别分化情况。共198株,其中雄株102株,雌株96株。经χ2检验,符合1: 1分离。从198个F1个体中随机挑选47株雄株和47株雌株共94个单株构成作图群体,进行作图分析。
1.3 叶片总DNA提取与检测2007年3月初采集鲜嫩叶片,采用改进的CTAB裂解-硅珠吸附法提取叶片总DNA(张博等,2004)。用1%的琼脂糖凝胶电泳检测DNA质量。
1.4 SSR标记分析SSR标记分析参照刘恩英等(2008)。反应体系(总体积为10 μL)为: Taq聚合酶0.5 U; Mg2+ 2.5mmol· L-1; dNTP各0.1 mmol·L-1; 引物(R & F)各0.3μmol·L-1; DNA 1 μL(20~50 ng·μL-1); 10×buffer(Tris-HCl 10 mmol· L-1 pH8.0,KCl 50 mmol·L-1)1μL; ddH2O补齐。PCR扩增程序为: 94 ℃ 2 min; 94 ℃ 45 s,(Tm -5)℃ 15 s,72 ℃ 45 s,35个循环; 72 ℃ 8 min; 4 ℃保温。PCR扩增产物采用8%变性聚丙烯酰氨凝胶(7 mol·L-1尿素)电泳(200 V稳压,1.4 h)分离,银染检测。
1.5 SRAP标记分析SRAP的总反应体系为10 μL,包括: Taq聚合酶0.75 U; Mg2+2.5 mmol·L-1; dNTP各0.2 mmol·L-1; 正反引物各0.3 μmol·L-1; DNA 1 μL(20~50ng·μL-1); 10×buffer(Tris-HCl 10 mmol·L-1 pH8.0,KCl 50 mmol·L-1)1 μL; ddH2O补齐。SRAP-PCR的扩增程序为: 94 ℃ 2 min; 94 ℃ 30 s,35 ℃ 30 s,72 ℃ 1 min,5个循环; 94 ℃ 30 s,50 ℃ 30 s,72 ℃1 min,35个循环; 72 ℃延伸8 min; 4 ℃保温。PCR产物经6%变性聚丙烯酰氨凝胶(7 mol·L-1尿素)电泳(200 V稳压,1.5 h)分离,银染检测。
1.6 遗传图谱的构建采用Joinmap 4.0构建遗传连锁图谱(van Ooijen,2006)。首先利用χ2检验检测标记偏分离,对于严重偏离期望比的标记在第1轮排序中排除,待其他标记排好后,再逐个插入。采用LOD≥3.0对标记分群,Kosambi函数计算图距,参考Hanley等(2002),设置连锁分析参数为: Rec=0.45,LOD=1.0,Jump=5。最后用MapChart 2.2绘制遗传图谱(Voorrips,2002)。
1.7 基因组长度的估算遗传图谱实际长度为框架图长度,即连锁群(>3个标记)长度之和; 连锁群总长度包括连锁群、三联体和二联体在内所有连锁长度的总和。参照Chakravarti等(1991)的方法估计遗传图谱长度,即框架图各连锁群实际长度乘(m+1)/(m-1)作为估计长度,m为每个连锁群的标记数。框架图覆盖率等于框架图长度占估算的遗传图谱总长度的百分比; 总的图谱覆盖率等于连锁群总长度占估算的遗传图谱总长度的百分比。
1.8 图谱比较利用本研究构建的图谱上的SSR标记的引物序列与BLAST工具(http://genome.jgi-psf.org/Poptr1_1/Poptr1_1.home.html),寻找毛果杨全基因组序列中2个引物序列之间的100~500 bp的序列,将其看作SSR标记,与之进行比较。若2个引物序列之间在毛果杨全基因组上难以寻找到100 ~500 bp的序列,就利用GenBank上包含该标记的柳树EST序列与毛果杨的全基因组序列进行比对(align),寻找毛果杨基因组中同源性超过75%的EST序列,视为SSR标记,与之比较。
2 结果与分析 2.1 引物筛选利用双亲和F1代的6个单株对1 656对杨树SSR引物进行筛选,其中637对引物有PCR扩增产物,引物通用率为38.47%。在F1群体中检测,获得66对多态性引物,多态性比例为3.99%(图 1)。在68对柳树SSR引物中有47对被成功扩增,引物通用率为69.12%。在F1群体中分析,获得19对多态性引物,多态性比例为27.94%。研究结果表明,SSR引物具有较高的特异性和保守性。

|
图 1 SSR标记O195在F1群体中的分离 Figure 1 The SSR-PCR product's segregation of the marker O195 in S. suchowensis × S. erioclada F1 family 箭头所指为多态位点; 分离类型: ab × cd; S:簸箕柳; E:绵毛柳。 Arrowheads indicate the polymorphic loci. Separate type is ab × cd. S is S. suchowensis; E is S. erioclada. |
对100个SRAP引物组合进行筛选,有52个引物组合谱带清晰,扩增谱带5~10条,最多26条,扩增片段在100~1 500 bp。经F1群体检测后,得到35个多态性引物组合,多态性比例为35%(图 2)。

|
图 2 SRAP标记在F1 Figure 2 Segregation of SRAP-PCR product in S. suchowensis × S. erioclada F1 family 引物组合: ME10SA4(箭头所指为多态位点); M:标准marker; S:簸箕柳; E:绵毛柳。 Primers: ME10SA4 (arrowheads indicate the polymorphic loci); M: Molecular mass marker; S: S. suchowensis; E: S. erioclada. |
筛选出的66对杨树SSR多态性引物,共产生82个分离位点,平均每对引物产生1.24个位点。筛选出的19对柳树SSR多态性引物,共产生24个分离位点,平均每对引物产生1.26个位点(图 1)。筛选出的35个SRAP多态性引物组合,能扩增出1~9条多态性谱带,平均每个组合产生3个多态位点(图 2)。2对柳树SCAR引物共扩增出8个多态位点; SCAR _ AE08-780扩增出6个多态位点,SCAR342-520扩增出2个多态位点。
利用χ2检测标记分离: ab×aa和aa×ab期望分离比为1 : 1(d=1),ab×ab期望分离比为1 : 2 : 1(d=2),ab×cd和ab×ac期望分离比为1 : 1 : 1 : 1(d=3)。经检测,共发现48个偏分离标记: 106个SSR标记中有11个偏离期望分离比(P<0.05),其中4个倾向父本,7个倾向母本,偏分离率为10.4%,其中严重偏离期望比(P<0.01)的有4个,占3.8%。11个SSR偏分离标记中只有1个是从柳树开发的,该标记偏向母本。8个SCAR标记中有2个偏离期望分离比。105个SRAP多态分离位点中有35个偏离期望分离比,其中18个偏向父本,17个偏向母本,偏分离率为33.3%,其中严重偏分离的有15个,占14.3%。
对标记信息来源的分析可以检验所选的群体是否适合遗传图谱的构建,如果群体偏向某一亲本,构建的图谱的可信度和应用范围会降低。本研究中,在106个SSR和8个SCAR多态位点中,有53个信息(46.5%)来自母本(ab×aa),54个(47.4%)来自父本(aa×ab),9个为双亲(ab×ab,ab×cd和ab×ac)共同提供; 在105个SRAP位点中,55个(52.4%)来自母本(ab×aa),50个(47.6%)来自父本(aa×ab)。可见来源于2个亲本的标记位点在整个群体中的分离比接近,没有偏向某一亲本的现象,这表明双亲为子代提供的遗传信息相当,所选作图群体适合构建柳树遗传图谱。
2.3 遗传图谱构建利用Joinmap4.0对获得的220个标记进行连锁分析,得到1张包含117个标记(56个SSR,54个SRAP,5个SCAR和2个性别标记)的遗传框架图。多于4个标记的连锁群有10个,另有12个三联体(triplet),11个二联体(doublet)。最大连锁群图距为178.1 cM,最小连锁图距为13.1 cM,总图距为1 631.4 cM; 标记间最大间距为42.3 cM,最小间距为1.4 cM,平均标记间距为14.1 cM(图 3)。

|
图 3 簸箕柳×绵毛柳遗传框架图 Figure 3 Genetic linkage map of S. suchowensis × S. erioclada 左侧为遗传图距,右侧为分子标记; 标记后缀数字:由该标记扩增出的片段从小到大的代码; 阴影:偏分离区域; d:偏分离,dd:严重偏分离; *s:偏母本的标记,*e:偏父本的标记。下同。 The left of the linkage group is genetic distances in centiMorgan and the right is marker names. Marker suffix numbers stand for a sequential number according to the size and beginning with the lowest size (in base pairs) of the DNA fragments scored. Shaded regions indicate blocks of distortion. Distorted markers note with the suffix "d" and "dd" which deviating at 0. 01 < P≤0. 05 and seriously deviating at P≤0. 01, respectively. *s indicates markers deviated toward female parent and *e toward male. The same below. |
48个偏分离标记中有20个被连锁于不同的连锁群上(图 3); 20个连锁到图谱的偏分离标记中包含15个SRAP、4个SSR和1个SCAR标记。有3个偏分离标记集中分布区域,分别在连锁群SE-2,SE-9和SE-18上; 区域内的标记都偏向于某一个亲本,SE-2和SE-18连锁群的8个偏分离标记偏向于父本,SE-9连锁群的2个偏分离标记偏向于母本。另外将性别分化性状标记(命名为雌性性别标记Sex-F和雄性性别标记Sex-M)分别被定位在第1和第17连锁群上(图 3)。
2.4 基因组长度估算根据Chakravarti等(1991)提出的估算方法,柳树基因组长度为2 261.39 cM,框架图覆盖率为60.29%,总的图谱覆盖率为72.14%。
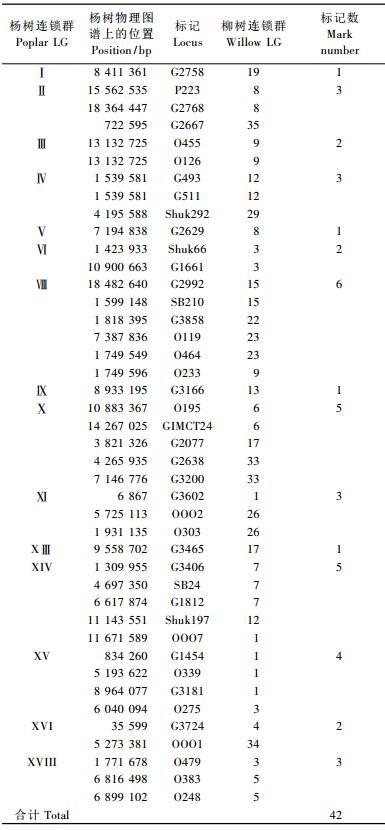
2.5 图谱比较通过与毛果杨全基因组序列比对发现,有来自杨树的36个SSR和柳树的10个SSR标记在毛果杨基因组中存在同源标记。46个同源标记中有42个分布在毛果杨的15个连锁群上,每个连锁群上的同源标记数为1~6个(表 1)。另外4对SSR标记O68,Sare5,P2020和O94分别在杨树的Scaffold _131,145,66和70上找到了同源标记(图 4)。
|
|

|
图 4 簸箕柳×绵毛柳遗传图与毛果杨物理图比对 Figure 4 An alignment of the S. suchowensis × S. erioclada linkage map to the P. trichocarpa physical map based on SSR markers ■表示毛果杨物理图,图中“O”表示连锁群起始端,“E”表示连锁群末端; □表示簸箕柳×绵毛柳遗传图。 ■ P. trichocarpa physical map, "O" indicates the beginning, "E" indicates the end. □ S. suchowensis × S. erioclada linkage map. |
根据Pelgas等(2006)2个连锁群为同源连锁群的标准: 1)至少有2个同源标记; 2)当只有1个同源标记时,视为推定同源连锁群(putativehomologous linkage groups)。本研究发现了5个同源连锁群(Ⅲ,Ⅵ,Ⅺ,ⅩⅤ和ⅩⅧ)和3个推定同源连锁群(Ⅰ,Ⅸ和ⅩⅢ),同时发现有2~4个亚连锁群(sub-group)与其他6个毛果杨的连锁群建立了同源关系,其中与LGⅧ建立同源关系的亚连锁群有4个,与LGⅩ,LGⅩⅣ建立同源关系的亚连锁群有3个,与LGⅡ,LGⅣ和LGⅩⅥ建立同源关系的亚连锁群分别有2个(图 4)。在毛果杨LGⅤ上的同源标记G2629与G2678及P223组成了与LGⅡ同源的连锁群(图 4中没有列出LGⅤ)。柳树同源连锁群SE-Ⅲ,SE-Ⅳ,SE-ⅩⅢ和SE-ⅩⅣ上分别有1个标记(即O233,Shuk197,G2077和G511)是LG Ⅷ,LGⅩⅣ,LGⅩ和LGⅣ连锁群上的同源标记; 同源连锁群SE-Ⅲ和SE-Ⅰ上分别有2个(O275和O479)和3个标记(G3602,OOO2和OOO7)是LGⅪ,LGⅩⅣ,LGⅩⅤ和LGⅩⅧ连锁群上的同源标记(表 1和图 4)。这些现象说明在杨柳科的进化过程中可能曾经发生过染色体的重排或复制事件,但也不能排除是由统计误差造成的。
总的来看,簸箕柳×绵毛柳和毛果杨的同源连锁群之间具有较好的标记共线性,42个同源标记中有36个具有同线性,占85.71%;36个具有同线性的标记中有34个具有共线性,占94.44%。
另外在Hanley等(2002)构建的蒿柳遗传图谱上,也发现3个同源标记(即SB24,SB210和P2020);3个同源标记中SB210和标记SB532及SB355组成了与毛果杨LGⅧ同源的连锁群(Hanley et al., 2006),与本研究结果相同; 但同源标记SB24和P2020所在的连锁群上的共显性标记信息太少,不易判断其同源关系。
3 讨论 3.1 标记对框架图的影响不同类型的分子标记具有各自的特点,构建遗传图谱时选用的分子标记要尽可能覆盖整个基因组,这样构建的连锁图谱才具有完整性和代表性,也才具有较高的实用价值。SSR标记为共显性标记,多态性好,重复性高,覆盖整个基因组,具有多等位基因的特性,是构建遗传连锁图谱较理想的分子标记。本研究筛选出的85对SSR多态性引物,共产生106个分离位点,平均每对引物产生1.25个位点; 共有56个SSR标记连锁到簸箕柳×绵毛柳的框架图上,分布于22个连锁群上,其中只有4个为偏分离标记。SRAP标记的正、反引物分别针对基因组的内含子和外显子区域,与SSR标记的扩增区域互补,可以作为SSR标记补充标记,有效地增加图谱的密度和基因组覆盖率,其多态性和效价比(产生多态性的效率/成本)都很高。本研究筛选出35个SRAP多态性引物组合,共产生105个多态位点,平均每对引物产生3个位点; 共54个SRAP标记连锁到框架图上,分布于23个连锁群上。
在杂交群体中检测到标记的偏分离现象是正常的(Grattapaglia et al., 1994)。在本研究中,SRAP标记偏分离的比率比SSR标记高,这是因为SRAP标记属于显性标记,显性标记比共显性更容易检测到偏分离(Gaudet et al., 2008)。造成杂交群体产生偏分离的原因有很多,本研究中发现的偏分离现象可能是由于配子体选择导致的,簸箕柳和绵毛柳因属于不同的组的2个种,存在一定的杂交不亲和现象,杂交后得到的种子经过自然选择只有一部分能萌发。不过,由于本研究分析的标记较少,对于偏分离的产生原因还需近一步的研究。
3.2 图谱特征本研究利用Joinmap4.0获得1张包含117个标记的簸箕柳×绵毛柳遗传框架图,是目前国内第1张柳树遗传图谱。构建图谱使用了多种分子标记,其中SRAP标记是首次被用于国内木本植物的图谱构建研究。获得的遗传框架图总图距为1 631.4 cM(图 3),在国外已发表的4张柳树图谱的图距范围之内(王源秀等,2008); 只是由于标记数相对较少,平均标记间距较大,为14.1 cM(图 3)。因此要想使本研究得到的遗传图谱在以后的柳树遗传学和基因组学研究中发挥更大的作用,则需要在该图谱上增加标记,包括一些显性标记,特别是通用共显性标记,以提高该图谱的密度和精度,也可为柳树遗传育种工作提供更可靠的基因组信息。
3.3 柳树的性别决定对于雌雄异花植物来说,不同性别的个体往往具有不同的特征,因此经济价值和育种策略也不同。大多数杨柳科树种为雌雄异花,其性别决定机制比较复杂。Yin等(2008)研究发现:杨树雌性个体中,性别决定位点附近区域的重组受到抑制、标记严重偏分离、染色单体异化、杂交后代性别严重偏向雄性,认为杨树中初步形成了雌性为杂合的ZW型性别染色体。Alstrom-Rapaport等(1998)研究发现蒿柳杂交后代雌性个体偏多,经RAPD标记分析发现标记UBC354560与雌性性别决定基因连锁,因此认为上位效应控制着标记与性别之间的关系。
本研究中2个SCAR标记(SCAR354-520和SCAR_AE08-780)的引物是来自NCBI数据库。标记SCAR354-520没有扩增出与性别共分离的位点,而SCAR_AE08-780-5是由标记SCAR_AE08-780扩增的1条长度约550~650 bp的片段,该标记位点在80.95%(37/48)的雌株(包括母本和47个F1子代个体)中和21.28%(10/47)的雄株中出现,与雌性性别标记Sex-F(相距29.7 cM)一起定位于图谱的第1连锁群(图 3)。可能第1连锁群与柳树的性别决定有关。不过,雄性性别标记Sex-M,被定位于第17连锁群上; 由于该图谱饱和度不高,不能判断第17连锁群和第1连锁群是否因为标记不足而被分开。在杨树遗传图谱研究中将性别分化位点都定位在第19同源连锁群的相近位置上(Yin et al., 2008; Zhang et al., 2009),而本研究的第1和17连锁群都不是杨树第19连锁群的同源连锁群,因此鉴于本研究的局限性,要判断柳树和杨树是否具有相同的性别决定机制,需要进一步的有效证据。
3.4 与毛果杨全基因组的比较本研究获得的是簸箕柳×绵毛柳遗传框架图,只能与毛果杨基因组进行初步比较研究。通过比较发现,与毛果杨基因组存在一定的标记同线性和共线性,在进化过程中具有较高的保守性; 共发现了46个同源标记、15个同源连锁群,85.71%的同源标记具有共线性(图 4)。比较中发现一些同源标记位置上的偏差(G2629,G2077,G 3602,OOO7,O275和O479),以及同一标记的多等位基因位点存在不同的同源连锁群上(OOO2,G511,O233和Shuk197)或同源连锁群的不同位置上(Sare5和G3181),这些现象说明在杨柳科的进化过程中可能曾经发生过染色体的重排或复制事件,也可能是由于基因型误差造成的,同样也可能由于比对误差的原因; 因为进行比对时,是直接将SSR引物序列或其原始EST序列与毛果杨全基因组进行比较,比较的结果仅仅代表引物本身或其原始EST在毛果杨染色体上的位置,但在实际PCR扩增时,其结果受到反应条件和引物保守性的影响,扩增出的柳树DNA片段很有可能不是与毛果杨基因组同源的目标位点。因此要进行详尽可信的杨柳科基因组图谱比较研究,一是需要增加标记,特别是杨柳科不同树种的通用共显性标记,这些标记位点可以作为锚定位点(constitute anchor points)构建密度和精度更高的柳树一致性图谱(consensus map)(Berlin et al., 2010); 二是需要将一些位点进行测序,特别是同一标记的直系(homolog)或旁系(paralog)同源基因位点(Pelgas et al., 2006),这样才能确定标记的同源性或确定不同树种在这些位点处是否出现重组。
邓玉营, 徐立安, 张博, 等. 2008. 黄花柳基因组微卫星分离及多态性位点检测[J]. 分子植物育种, 6(1): 89-94. |
刘恩英, 王源秀, 孙晨晨, 等. 2008. 柳树SSR反应体系的建立与优化[J]. 江苏林业科技, 35(5): 8-11. |
涂忠虞. 1982. 柳树育种与栽培[M]. 南京: 江苏科学技术出版社.
|
王源秀, 徐立安, 黄敏仁. 2008. 柳树遗传学研究现状与前景[J]. 植物学通报, 25(2): 240-247. |
杨琦, 张鲁刚. 2007. 大白菜SRAP反应体系的建立与优化[J]. 西北农业学报, 16(3): 119-123. |
张博, 张露, 诸葛强, 等. 2004. 一种高效的树木中DNA提取方法[J]. 南京林业大学学报:自然科学版, 28(1): 13-16. |
Alstrom-Rapaport C, Lascoux M, Wang Y C, et al. 1998. Identification of a RAPD marker linked to sex determination in the basket willow(Salix viminalis L)[J]. J Hered, 89(1): 44-49. DOI:10.1093/jhered/89.1.44 |
Barcaccia G, Meneghett S, Albertini E, et al. 2003. Linkage mapping in tetraploid willow: segregation of molecular markers and estimation of linkage phase support an allotetraploid structure for Salix alba×Salix fragilis interspecific hybrids[J]. Heredity, 90(2): 169-180. DOI:10.1038/sj.hdy.6800213 |
Berlin S, Lagercrantz U, von Arnold S, et al. 2010. High-density linkage mapping and evolution of paralogs and orthologs in Salix and Populus[J]. BMC Genomics, 11: 129. DOI:10.1186/1471-2164-11-129 |
Chakravarti A, Lasher L K, Reefer J E. 1991. A maximum likelihood for estimating genome length using genetic linkage data[J]. Genetics, 128(1): 175-182. |
Grattapaglia D, Sederoff R. 1994. Genetic linkage maps of Eucalyptus grandis and Eucalyptus urophylla using a pseudo-testcross mapping strategy and RAPD markers[J]. Genetics, 137(4): 1121-1137. |
Gaudet M, Jorge V, Paolucci I, et al. 2008. Genetic linkage maps of Populus nigra L.including AFLPs, SSRs, SNPs, and sex trait[J]. Tree Genet Genom, 4(1): 25-36. |
Hanley S J, Barker J H, van Ooijen J W, et al. 2002. A genetic linkage map of willow(Salix viminalis)based on AFLP and microsatellite markers[J]. Theor Appl Genet, 105(6/7): 1087-1096. |
Hanley S J, Mallott M D, Karp A. 2006. Alignment of a Salix linkage map to the Populus genomic sequence reveals macrosynteny between willow and poplar genomes[J]. Tree Genet Genom, 3(1): 35-48. DOI:10.1007/s11295-006-0049-x |
Pelgas B, Bezuseigle S, Acheré V, et al. 2006. Comparative genome mapping among Picea glauca, P.mariana×P.rubens and P.abies, and correspondence with other Pinaceae[J]. Theor Appl Genet, 113(8): 1371-1393. DOI:10.1007/s00122-006-0354-7 |
Rǒnnberg-Wǎstljung A C, Tsarouhas V, Semerikov V, et al. 2003. A genetic linkage map of a tetraploid Salix viminalis×S.dasyclados[J]. hybrid based on AFLP markers.Forest Genet, 10(3): 185-194. |
Tsarouhas V, Gullberg V, Lagercrantz V. 2002. An AFLP and RFLP linkage map and quantitative trait locus(QTL)analysis of growth traits in Salix[J]. Theor Appl Genet, 105(2/3): 277-288. |
Tuskan G A, Jansson S, Bohlmann J, et al. 2006. The genome of black cottonwood, Populus trichocarpa(Tort. & Gray)[J]. Science, 313(5793): 1596-1604. DOI:10.1126/science.1128691 |
van Ooijen J W. 2006. Joinmap 4, Software for the calculation of genetic linkage maps in experimental populations.Kyazma BV, Wageningen[J]. Netherlands. |
Voorrips R E. 2002. MapChart: Software for the graphical presentation of linkage maps and QTLs[J]. J Hered, 93(1): 77-78. DOI:10.1093/jhered/93.1.77 |
Yin T M, DiFazio S P, Gunte L E, et al. 2008. Genome structure and emerging evidence of an incipient sex chromosome in Populus[J]. Genome Res, 18(3): 422-430. DOI:10.1101/gr.7076308 |
Zhang B, Tong C, Yin T M, et al. 2009. Detection of quantitative trait loci influencing growth trajectories of adventitious roots in Populus using functional mapping[J]. Tree Genet Genom, 5(3): 539-552. DOI:10.1007/s11295-009-0207-z |