 2011, Vol. 47
2011, Vol. 47

2. Key Laboratory of Forest Genetics and Gene Engineering, Nanjing Forest University Nanjing 210037
2. 南京林业大学林木遗传和基因工程重点实验室 南京 210037
Poplar is a fast-growing, widely-cultivated tree species and is one of the major woody plants used for industrial plantation in China. Poplars are widely planted in various regions of China. At present, poplar plantations are primarily established by cloned ramets. However, in some regions, plant diseases and insect pests pose serious threats to poplar plantations; among these, poplar black spot disease caused by pathogen Marssonina brunnea is rampant and spreads easily throughout the plantation, which causes premature defoliation and affects the photosynthetic efficiency, resulting in seriously stunt tree growth (Han et al., 1998).
Intensive studies have been made in recent years for specific resistance to rust in poplars based on genetic maps, with emphasis on resistance to Melampsora×columbiana and M. larici-populina races E1, E2 and E3 (Stirling et al., 2001; Zhang et al., 2001; Yin et al., 2004). Rust resistance between the host and pathogen is very specific and fit for the category of Flor's gene for gene hypothesis (Flor, 1971). However, pathogen Marssonina has a wide range of hosts, and our early studies showed that the resistance of poplar to this tribe of pathogens was in a horizontal manner, thus could involve a much more complex molecular mechanism. The horizontal resistance is a result of coordinated action of many resistance-associated genes. Therefore, when studying the function and relationships between such genes, the entire genome should be considered. The accomplishment of whole poplar genome sequencing showed that the NBS-LRR resistance genes in poplar were more abundant than in Arabidopsis, and the significantly different aggregation and unevenly distribution of resistance genes on different chromosome indicated different chromosome sustained different selection pressure by pathogens(Yin et al., 2008).Large-scale expressed sequence tags(ESTs) sequencing provides a gateway into the genome of an organism; ESTs and databases have proved to be powerful tools for gene discovery, gene mapping, and for the analysis of quantitative traits (Luo et al., 2005; Sterky et al., 2004).
Poplar is a model system for genome studies in perennial plant species, which possess a relatively small genome with identical chromosome numbers among different poplar species (2n=38) (Bradshaw, 2000; Taylor, 2002; Bhalerao, 2003). It is the first tree that the whole genome sequence has been accomplished (Tuskan et al., 2006). In addition, the poplar EST project has thus far been quite successful, and large amounts of ESTs have been deposited to GenBank database (Sterky et al., 2004; http://www.ncbi.nlm.nih.gov). The poplar genomic resources provide major new study opportunities in plants.
To understand the mechanism of horizontal resistance caused by pathogen Marssonina brunnea in poplar, the EST approach was used to analyze changes in gene expression. In this study, we inoculated poplar leaves of the resistant Populus deltoides 'Lus' (I-69/55) and susceptible P. euramericana 'I-45/51' with the black spot disease pathogen Marssonina brunnea and built the cDNA library correspondingly, then performed cDNA sequencing and functional ontology. By alignment analysis with Populus trichocarpa assembly v1.0 (http://genome.jgi-psf.org/Poptr1), some resistance-associated candidate genes were classified and located on the linkage groups of Populus genome. This study supplied a resource of candidate genes for further exploring the genetic mechanism for the host horizontal resistance to pathogen Marssonina brunnea at whole genome range and provided important information for further gene discovery.
1 Materials and Methods 1.1 Plant materialsWe inoculated Populus deltoides 'Lus' (I-69/55) clone that is resistant to black spot disease, and the P. euramericana 'I-45/51' clone that is susceptible to black spot disease with Marssonina brunnea f. sp. brunnea (the strain was provided by pathology group of Nanjing Forestry University). The poplar material cuttings were planted in greenhouse, and when several young leaves spread out, the pathogen was inoculated. The young leaf tissues at seventy-two hours after inoculation were harvested for using cDNA library construction and ESTs sequencing. The samples were cooled quickly in liquid nitrogen, and subsequently stored at -80 ℃.
1.2 cDNA libraries construction and EST sequencingcDNA libraries L45-72 and L69-72 were constructed with ZAP Express cDNA synthesizing kits (Stratagene) and ZAP Express cDNA Gigapack Ⅲ gold cloning kits (Stratagene), respectively(Zeng et al., 2004).
Plasmid DNA was extracted with 96 easy plasmid DNA preparation kits produced by Vitagene Biotechnologies Co. Ltd. The sequencing primer was the universal primer T3 (5′-ATTAACXCCTCACTAAAGGGA-3′) that binds to both sides of the multiclonal site of the λZAP vector. For 5′-terminal sequencing, cDNA clones were randomly selected. The Automatic DNA Analyzer 3100 (ABI) was used for sequencing, and the sequencing kits were BigDye Terminater v3.1. The reaction was conducted in a 10 μL volume, including BigDye (0.5 μL), 5× sequencing Buffer (1.75 μL), sequencing primer (1 μL, 3.2 pmol·μL-1), and sequencing templates (150-300 ng), with the remaining volume made up with sterilized water. PCR sequencing condition was as following: 96 ℃ for 2 minutes; 94 ℃ for 10 seconds; 51 ℃ for 5 seconds; 60 ℃ for 4 minutes (repeated sequence for 25 cycles), followed by 4 ℃ incubation. To purify the PCR products, they were precipitated in NaAc (pH 5.2) and 95% ethanol, centrifuged and then the supernatants were removed and discarded. Resulting sediments were washed with 70% ethanol, the supernatants were discarded and the samples were dried. Final sediments were dissolved in Hi-DiTM formamide (ABI) and were denatured at 95 ℃ for 4 minutes. After cooling quickly to room temperature, the sediments were subjected to sequencing on an ABI sequencer 3100 or were stored at -20 ℃ for later use.
1.3 EST clustering and annotationSequence clustering was performed with the Phred/Phrap/Consed (Ewing et al., 1998a; 1998b) software. Low-quality sequences at both sides of the target sequences were removed by Phred (Q13 criteria), then the cross-match program was used to screen vector sequences. Finally, Phrap was used to splice valid sequences that were longer than 100 bp.
The clustered sequences were subjected to sequence similarity comparison within the GenBank Nt Database, GenBank Nr Database, and SWISSPROT Database using BLAST software (Altschul et al., 1997). Functional annotation was based on the optimal sequence. The parameters were selected as follows: the limits to Nt comparison results were E value < 1e-15 and similarity >30%; the limits to Nr and SWISSPROT comparison results were E value < 1e-10 and similarity >25%. Subsequently, according to SWISSPROT ACCESSION, tentative unigenes were functionally classified by Gene Ontology (http://www.geneontology.org/).
1.4 Analysis of resistance-associated candidate genesThe sequences of tentative unigenes associated to plant resistance based on the annotation from GenBank Nr Database were selected out and made BLAST alignment with Populus trichocarpa masked assembly v1.0 (http://genome.jgi-psf.org/Poptr1) with expect 1e-20. Then, according to the corresponding determinate location on Populus genome, corresponding Populus gene and functional annotation, the resistance-associated candidate genes were classified and located on the linkage groups of Populus genome.
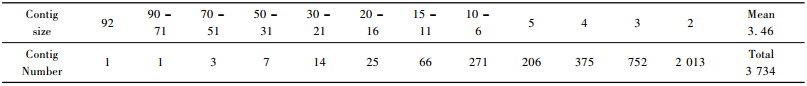
2 Results 2.1 EST sequencing and clusteringIn this study we used two cDNA libraries, L45-72 and L69-72 (Zeng et al., 2004). From the two cDNA libraries of poplar leaves, clones were randomly chosen for 5′ sequencing, and a total of 21 657 EST sequences were obtained. After eliminating low-quality or vector sequences, 20 023 sequences with a valid length of more than 100 bp (mean length 615.9 bp) were obtained. There were 9 866 valid EST sequences in the L69-72 library, the GenBank accession numbers ranged from CX167465 to CX177330. Whereas in the L45-72 library, there were 10 157 valid EST sequences, the corresponding GenBank accession numbers ranged from CX177331 to CX187487. At the P-value cut-off 0.01, all the quality sequences were trimmed off with Q13 (95% accuracy) and were clustered, these 20 023 valid sequences yielded a total of 10 816 tentative unigenes, including 3 734 contigs and 7 082 singletons, with a primary unigene sequence length between 500 and 800 bp (Tab. 1). Of these 10 816 tentative unigenes, there were 4 659 unigenes in which the ESTs were only from L45-72, 5 238 unigenes in which ESTs were only from L69-72, and the other 919 unigenes in which the ESTs from the both libraries. Of the 3 734 contigs sequences, the maximum contig size (the number of EST sequences spliced into contig) was 92, and the minimum size was 2 (Tab. 2). Among them, the longest contig sequence was 3 499 bp (consisted of 34 EST sequences).
|
|

|
|
The result of Blast alignment within GenBank non-redundant nucleic acid and protein databases indicated that there were 8 853 tentative unigenes homologous to genes in the database (annotated), and 1 963 tentative unigenes that were non-homologous to genes in the database (not annotated). The latter may reflect new genes that have not yet been annotated. After a 72 h inoculation with black spot disease pathogen Marssonina brunnea f. sp. brunnea, we performed functional interpretation and classification analysis of all tentative unigenes. As shown in Tab. 3, all tentative unigenes were classified according to the three classification components, namely molecular function, biological process and cellular component, as formulated by Gene Ontology (http://www.geneontology.org). These genes covered a broad range of the GO functional categories. However, due to the lack of gene products information, many transcripts can not be functionally categorized. We successfully classified 10 717 tentative unigenes in terms of biological processes, 9 741 tentative unigenes in terms of molecular function, and 7 127 tentative unigenes in terms of cellular components. Since one gene product may be assigned to more than one GO term, and one child term can fit into multiple parental categories, the total number of GO mappings in the three ontologies will exceed the number of all the tentative unigenes.
|
|

Among the 9 741 tentative unigenes by molecular function, 3 386 appeared to be of catalytic function and 2 293 contributed to binding, while others contribute to antioxidant, signal transduction, defense and immunity, apoptosis regulation and transcription regulation, all of which were evidently associated with resistance and stress response. Comparing the functional genes from resistant and susceptible cDNA libraries, the differentially expressed gene in the L69-72 mainly included bZip family transcription factor, D-3-phosphglycerate deghdrogenase, hydroxyproline-rich glycoprotein family protein, phenylalanine ammonia-lyase, acetohydroxy acid isomeroreductase, CuZn-superoxide dismutase, epoxide hydrolase, protein kinase and expressed protein and so on. The differentially expressed gene in the L45-72 mainly involved ethylene response factor, leucine-rich repeat family protein/protein kinase family protein, pathogenesis-related protein, chalcone synthase, chitinase, beta-1, 3-glucanase, peroxidase, polyubiquitin, metallothionein, extension protein, zinc finger family protein and so on.
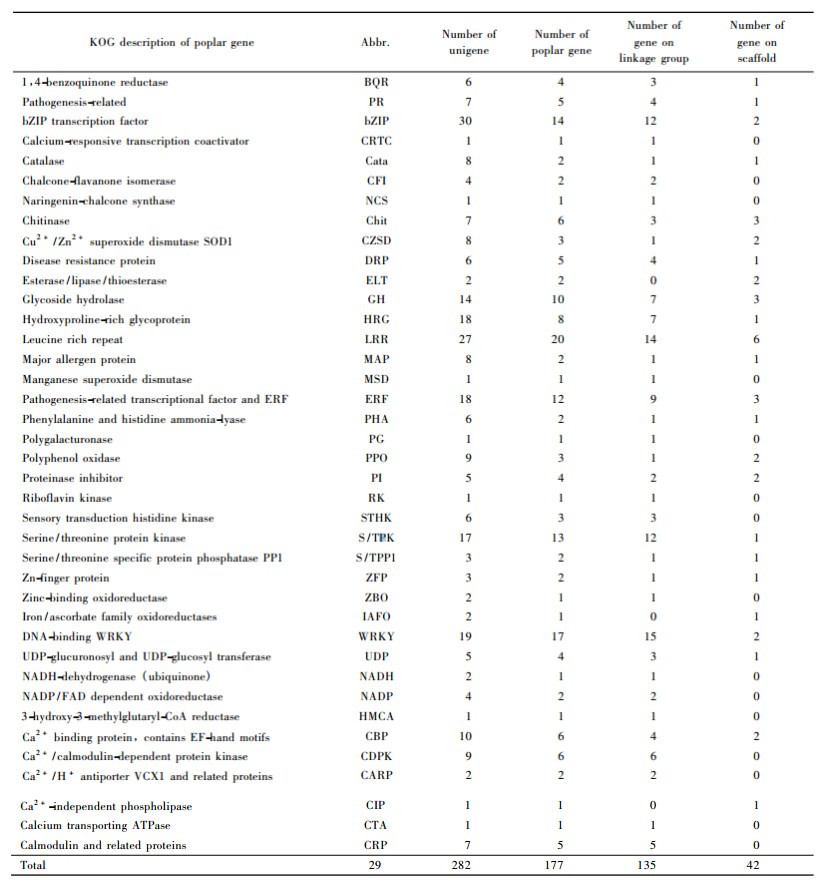
2.3 Resistance-associated candidate gene finding and localizationIn this study, all the 20 023 cDNA sequences were sequenced from the two poplar leave libraries inoculated by pathogen Marssonina brunnea f. sp. brunnea. To find some resistance-associated candidate genes, 330 unigenes (composed by 639 ESTs) which is functionally relevant to plant resistance were selected out from 8 853 annotated tentative unigenes, after the BLAST alignment with Populus trichocarpa masked assembly v1.0 (http://genome.jgi-psf.org/Poptr1) with expect 1e-20, 177 resistance-associated candidate Populus genes were found and they presented in 282 tentative unigenes (composed by 554 EST), the other 48 tentative unigenes were homologous with other Populus genes with different functions. The 177 candidate genes were classified into 39 different functional categories (Tab. 4). Among these genes, there were 135 genes located on the different linkage groups of Populus genome, and 42 genes located on the different scaffolds (Tab. 4). In order to clarify the determinate location and function of these 135 candidate genes on Populus genome, these genes were mapped on 19 linkage groups in name of their abbreviation (Tab. 4) and their start position by MapChart software (Voorrips, 2002) (Fig. 1). The results showed that all these candidate genes were not scattered evenly on different linkage groups, there were more than 10 candidate genes on LG_Ⅰ (16), LG_Ⅱ (14), LG_Ⅲ (13), LG_Ⅹ (10) and LG_XVI (10), but there was only one gene on LG_XVII, and there was no gene found on the LG_XV. As for the number of different functional gene we found, there was a big difference among them. The biggest gene group we found was leucine rich repeat which included 20 genes, the next was transcriptional factor WRKY which included 17 genes, the other big gene groups included bZIP transcription factor (14), Serine/threonine protein kinase (13), Pathogenesis-related transcriptional factor and Ethylene reaction factor (12) and Glycoside hydrolase (10). The small gene groups we found mostly included 1 or 2 genes, but all of them played a very important role in the function of plant resistant disease.
|
|

|
Fig.1 The map of resistance-associated candidate genes on Populus linkage groups |
EST is a quick and economical method for discovery of genes with moderate to abundant transcript levels. The relative abundance of the mRNA in a tissue is approximately reflected in the abundance of its corresponding cDNA in non-normalized libraries. Random sequencing of cDNAs, therefore, yields information about the relative expression levels of the genes represented by the EST (Adams et al., 1993; Covitz et al., 1998). In the study of comparison of expression profiles based on maize EST frequency analysis (Fernandes et al., 2002), 73 000 EST representing multiple organs and developmental stages of maize identified approximately 22 000 tentative unigenes in which singletons comprised 51.4% and 27.0% of the contigs contained only two or three EST. In the present study, clustering analysis of valid EST sequences indicated that the majority of tentative unigenes were also accounted for by singletons and by contigs with two or three EST. Of all 10 816 tentative unigenes, 7 082 singletons accounted for 65.5%; there were 2 765 contigs that contained two or three EST, which accounted for 25.6%. These EST should represent moderately expressed genes in the particular tissue source, because EST of rarely expressed genes are unlikely to be sampled. The unigenes that contained more than three EST presumably represent genes that are highly expressed in particular tissues.Knowledge of EST representation provides candidate genes for recovery of promoters conferring stage or organ-specific expression. (Fernandes et al., 2002).
Because a gene can be expressed as mRNA many times, EST ultimately derived from this mRNA may be redundant. In this study, we sequenced 20 023 EST which clustered into 10 816 non-redundant tentative unigenes. This corresponded to a redundancy of 46.0%. The result of gene discovery in the wood-forming tissues of poplar (Sterky et al., 1998), showed that a total of 751 clusters and 2 237 singletons were formed after assembly of the 4 809 EST from the cambial region (37.9% redundancy), the 883 developing xylem EST were assembled into 78 clusters and 653 singletons (17.2% redundancy). In the study of poplar root transcriptome (Kohler et al., 2003), 7 013 EST were assembled into 1 347 clusters and 3 527 singletons, the redundancy corresponded to 30.5%. All these studies indicated that the redundancy in different study was related to the number of EST analyzed and the cDNA library constructed. A normalized cDNA library can be used to reduce the redundancy and to maximize the chances of finding new genes through random sequencing (Soares et al., 1994), but the EST from the two non-normalized cDNA libraries basically represented the original gene expression abundance in this study.
3.2 Plant resistance to diseasePlant disease resistance and its underlying mechanisms have always been the focus of phyto-pathologic and plant breeding research. After being invaded by pathogens, plants most commonly display hypersensitivity responses that trigger a series of defensive responses and result in tissue or systematic resistance. There are two common molecular mechanisms underlying disease-resistance: hypersensitivity responses(HR) (Baker et al., 1997) and systematic acquired resistance (SAR) (Ryals et al., 1995). In each case, the disease-resistant function is carried out by the products of so-called "defensive" genes, and the cascades inducing gene expression are similar in both cases and involve signal recognition, signal transduction, and expression of genes. In the present study, on the basis of EST sequencing, we identified the expression of some genes associated with resistance to black spot disease in poplar.
Numerous studies have shown that the reception and transduction of pathogen signals by plant hosts are similar to those used by animal hosts, and that such processes require second messengers and protein phosphorylation. Ca2+, a second messenger participating in the growth and development of plants, is increasingly believed to participate in plant responses to adverse conditions (Yang et al., 2002; Pandey et al., 2002; Olmos et al., 2003). For example, various stimuli, such as light, adverse environment, pathogen infection, and phylohormones, will lead to signal transduction via changes in cytoplasmic Ca2+ concentration in plants. However, further transmission of Ca2+ signals requires Ca2+-binding proteins and Ca2+-dependent kinases. The Ca2+ messenger system plays a vital role in regulating gene expression, protease activity, and active oxygen production (Yang et al., 2002; Luan et al., 2002; Kim et al., 2002; Zhang et al., 2003). In addition, phosphorylation and dephosphorylation are intimately involved in signal recognition and transduction in plant defensive responses (Romeis, 2001). Factors closely associated with adverse signals mainly include mitogen-activated protein kinase (MAPK), Ca2+-dependent but calmodulin-independent protein kinase (CDPK), and receptor protein kinase (RPK). MAPK and CDPK can be activated by pathogen excitation (Treisman, 1996), and can regulate the expression of genes associated with disease resistance (Frye et al., 2000). In the present study, we found several kinds of Ca2+ signal related proteins, including 30 tentative unigenes with high sequence frequencies corresponding to 21 poplar genes (Tab. 4), which suggest that Ca2+ signals play a key role in the signal transduction system and in preventing infection by black spot disease pathogen in poplar.
Recent studies have shown that salicylic acid is an important component of the signal transduction process that not only activates a series of defensive responses after pathogen infection, and also necessary for a hypersensitivity response and systemic acquired resistance (Hammond-Kosack et al., 1996; van Wees et al., 2000; Chaman et al., 2003). Phenylalanine ammonia-lyase, which is a key enzyme in the phenylalanine metabolic pathway, not only controls the efficiency of phenylalanine, but is also involved in salicylic acid synthesis. Salicylic acid is a secondary metabolite in plants, and thus cannot be measured directly by gene transcription. However, according to levels of the key enzymes of its synthetic pathway and the gene expression of downstream responses, the role of the salicylic acid signal system in resistance to poplar black spot disease can be roughly determined. Indeed, in the present study we detected 6 tentative unigenes of phenylalanine and histidine ammonia-lyase as well as the presence of multiple salicylic acid binding proteins. In addition, we obtained 6 sequences associated with jasmonic acid function and many sequences associated with ethylene function. Nevertheless, further evidence is needed to clarify the role of salicylic acid, jasmonic acid and ethylene in resistance to poplar black spot disease.
In plant genomes, a large proportion of genes contribute to environmental change-associated signal transduction or transcription regulation, and these genes frequently appear in families. For instance, there are approximately 1 500 transcription factors in Arabidopsis thaliana, and they all belong to various gene families (Riechmann et al., 2000). The ethylene-responsive-element-binding factors (EREBF) family plays a role in responding to low temperature, drought, pathogens and pathogen inducers (Singh et al., 2002); the basic leucine-zipper (bZIP) family, in contrast, contributes to defensive response to pathogens and adverse stimuli (Jakoby et al., 2002). The WRKY family, a family of recently-discovered plant-specific transcriptional regulatory factors from many types of plants, is primarily involved in the defensive response to pathogens (Dellagi et al., 2000; Maleck et al., 2000). Of the ESTs obtained in this study, many have been reported to participate in regulating gene expression as transcription factors, in which WRKY transcription factors corresponded to 17 poplar genes, bZIP corresponded to 14 poplar genes and EREBF corresponded to 12 poplar genes, as well as a number of zinc-finger proteins. These transcription factors were expressed frequently in the libraries of poplar leaves induced by black spot disease pathogen, and were thought to contribute to the regulation of the expression of resistance genes.
During the interaction between plants and pathogen, many secondary metabolites take part in resistance and defensive responses (Hadacek, 2002). In the present study, the phenylalanine ammonia-lyase was highly expressed after pathogen exposure, suggesting that during the pathogenesis of black spot disease, lignin was synthesized in poplar leaves in large quantities. Other important type of genes such as oxidoreductases, catalase, chitinase, superoxide dismutase and so on all contributed to disease resistance and defensive response to poplar black spot disease.
The results in this study are indicative of changes to multiple metabolic pathways during the pathogenesis of black spot disease in poplar, and these changes clearly involve many genes and complex mechanisms. And the analyses of known functional genes indicated that a large proportion of expressed sequences were related to the plants resistance to disease or adverse environments. According to the structural characteristics of plant resistance genes, the Leucine rich repeat(LRR), Serine/threonine protein kinase, bZIP and WRKY transcription factors were involved; according to the role of signal molecules in systematic plant resistance responses, key signal molecules believed to be involved are salicylic acid, jasmonic acid and ethylene; and according to the categories of pathogenesis-related (PR) proteins induced systematically in plant defensive responses, this study identified a possible role for enzymes involved in degrading pathogen cell wall, such as chitinase, β-1, 3-glucanase, enzymes that inhibit pathogen growth, such as reductase, oxidoreductase, superoxide dismutase, and proteinase inhibitors. These findings demonstrated that the resistance to black spot disease in poplar is a complex network involving co-expression of many genes, and is closely related to other biological or non-biological adverse factors. Meanwhile, these results suggested that information about tissue-specific gene expression in poplar leaves exposed to pathogen of black spot disease is critical to our understanding of poplar resistance in cases of disease exposure as well as exposure to other stressful factors.
Using high throughput detection methods, such as cDNA microarrays based on these EST information or gene chips based on the whole poplar genome produced by Affymetrix or Roche NimbleGen to study the whole level gene expression of poplar varieties or species may shed light on the interrelationship between various resistance responses and aid in the discovery of genes that regulate various resistance responses, thus laying a foundation for further study of the mechanisms of plant disease resistance. In our next step of work, microarray and RT-PCR methods will be used for the gene expression analysis of different inoculated stage and candidate gene validation of disease resistance in poplar.
Adams M D, Kerlavage A R, Fields C, et al. 1993. 3400 new expressed sequence tags identify diversity of transcripts in human brain[J]. Nat Genet, 4(3): 256-267. DOI:10.1038/ng0793-256 |
Altschul S F, Madden T L, Schaffer A A, et al. 1997. Gapped BLAST and PSI-BLAST:a new generation of protein database search programs[J]. Nucl Acid Res, 25(17): 3389-3402. DOI:10.1093/nar/25.17.3389 |
Baker B, Zambryski P, Staskawicz B, et al. 1997. Signaling in plant-microbe interactions[J]. Science, 276(5313): 726-733. DOI:10.1126/science.276.5313.726 |
Bhalerao R. 2003. Out of the woods:forest biotechnology enters the genomic era[J]. Curr Opin Biotechno, 14(2): 206-213. DOI:10.1016/S0958-1669(03)00029-6 |
Bradshaw H D. 2000. Emerging model systems in plant biology:poplar as a model forest tree[J]. J Plant Growth Regu, 19(3): 306-313. DOI:10.1007/s003440000030 |
Chaman M E, Copaja S V, Argandona V H. 2003. Relationships between salicylic acid content, phenylalanine ammonia-lyase (PAL) activity and resistance of Barley to aphid infestation[J]. J Agric Food Chem, 51(8): 2227-2231. DOI:10.1021/jf020953b |
Covitz P A, Smith L S, Long S R. 1998. Expressed sequence tags from a root-hair-enriched Medicago trancatula cDNA library[J]. Plant Physiol, 117(4): 1325-1332. DOI:10.1104/pp.117.4.1325 |
Dellagi A, Helibronn J, Avrova A O. 2000. A potato gene encoding a WRKY-like transcription factor is induced in interactions with Erwinia carotovora subsp[J]. astroseptica and phytophthora infestans and is coregulated with classⅠendochitinase expression. Mol Plant Microbe Interact, 13(10): 1092-1101. |
Ewing B, Green P. 1998a. Base-calling of automated sequencer traces using phred. Ⅱ. Error probabilities[J]. Genome Res, 8(3): 186-194. DOI:10.1101/gr.8.3.186 |
Ewing B, Hillier L, Wendl M C, et al. 1998b. Base-calling of automated sequencer traces using phred.Ⅰ. accuracy assessment[J]. Genome Res, 8(3): 175-185. DOI:10.1101/gr.8.3.175 |
Fernandes J, Brendel V, Gai X W, et al. 2002. Comparison of RNA expression profile based on maize expressed sequence tag frequency analysis and micro-array hybridization[J]. Plant Physiol, 128(3): 896-910. DOI:10.1104/pp.010681 |
Flor H H. 1971. Current status of the gene-for-gene concept[J]. Annu Rev Phytopathol, 9: 275-296. DOI:10.1146/annurev.py.09.090171.001423 |
Frye C A, Tang D Z, Innes R W. 2000. Negative regulation of defense response in plants by a conserved MAPKK kinase[J]. P Natl Acad Sci USA, 98(3): 373-378. |
Hadacek F. 2002. Secondary metabolites as plant traits:current assessment and future perspectives[J]. Crit Rev Plant Sci, 21(3): 273-322. |
Hammond-Kosack K E, Jones J D. 1996. Resistance gene-dependent plant defense response[J]. Plant Cell, 8(10): 1773-1791. DOI:10.1105/tpc.8.10.1773 |
Han Zhengmin(韩正敏), Li Chuandao(李传道), Huang Minren(黄敏仁). 1998. Comparative studies of isolates of Marssonina brunnea in China. Scientia Silvae Sinicae (林业科学), 34(3): 59-65.
|
Jakoby M, Weisshaar B, Drge-Laser W, et al. 2002. bZip transcription factors in Arabidopsis[J]. Trends Plant Sci, 7(3): 106-111. DOI:10.1016/S1360-1385(01)02223-3 |
Kim M C, Panstruga R, Elliott C, et al. 2002. Calmodulin interacts with MLO protein to regulate defense against mildew in barley[J]. Nature, 416: 447-451. DOI:10.1038/416447a |
Kohler A, Delaruelle C, Martin D, et al. 2003. The poplar root transcriptome:Analysis of 7000 expressed sequence tags[J]. FEBS Lett, 542: 37-41. DOI:10.1016/S0014-5793(03)00334-X |
Luan S, Kudla J, Rodriguez-Concepcion M, et al. 2002. Calmodulins and calcineurin B-like proteins calcium sensors for specific signal response coupling in plants[J]. Plant Cell, 14(Supplement 1): S389-S400. |
Luo M, Dang P, Guo B Z, et al. 2005. Generation of expressed sequence tags for gene discoery and marker development in cultivated peanut[J]. Crop Sci, 45: 346-353. DOI:10.2135/cropsci2005.0346 |
Maleck K, Levine A, Eulgem T, et al. 2000. The transcriptome of Arabidopsis thaliana during systemic acquired resistance[J]. Nat Genet, 26: 403-410. DOI:10.1038/82521 |
Olmos E, Martinez-Solano J R, Piqueras A, et al. 2003. Early steps in the oxidative burst induced by cadmium in cultured tabacco cells (BY-2 line)[J]. J Exp Bot, 54(381): 291-301. DOI:10.1093/jxb/erg028 |
Pandey S, Tiwari S B, Tyagi W. 2002. A Ca2+/CaM-dependent kinase from pea is stress regulated and in vitro phosphorylates a protein that binds to AtCaM5 promoter[J]. Eur J Bioche, 269(13): 3194-3204. |
Riechmann J L, Heard J, Martin G, et al. 2000. Arabidopsis transcription factors:genome-wide comparative analysis among eukaryotes[J]. Science, 290(5499): 2105-2110. DOI:10.1126/science.290.5499.2105 |
Romeis T. 2001. Protein kinases in the plant defence response[J]. Curr Opin Plant Biol, 4(5): 407-414. DOI:10.1016/S1369-5266(00)00193-X |
Ryals J, Lawton K A, Delaney T P, et al. 1995. Signal transduction in systemic acquired resistance[J]. P Natl Acad Sci USA, 92(10): 4202-4205. DOI:10.1073/pnas.92.10.4202 |
Singh K B, Foley R C. 2002. Transcription factors in plant defense and stress responses[J]. Curr Opin Plant Biol, 5(5): 430-436. DOI:10.1016/S1369-5266(02)00289-3 |
Soares M B, Bonaldo M F, Jelene P, et al. 1994. Construction and characterization of a normalized cDNA library[J]. P Natl Acad Sci USA, 91: 9228-9232. DOI:10.1073/pnas.91.20.9228 |
Sterky F, Bhalero R R, Unneberg P, et al. 2004. A Populus EST resource for plant functional genomics[J]. P Natl Acad Sci USA, 101(38): 13951-13956. DOI:10.1073/pnas.0401641101 |
Sterky F, Regan S, Karlsson J, et al. 1998. Gene discovery in the wood-forming tissues of poplar:analysis of 5692 expressed sequence tags[J]. P Natl Acad Sci USA, 95(22): 13330-13335. DOI:10.1073/pnas.95.22.13330 |
Stirling B, Newcombe G, Vrebalov J, et al. 2001. Suppressed recombination around the MXC3 locus, a major gene for resistance to poplar leaf rust[J]. Theor Appl Genet, 103: 1129-1137. DOI:10.1007/s001220100721 |
Taylor G. 2002. Populus:Arabidopsis for forestry. Do we need a model tree?[J]. Ann Bot, 90(6): 681-689. DOI:10.1093/aob/mcf255 |
Treisman R. 1996. Regulation of transcription by MAP kinase cascades[J]. Curr Opin Cell Biol, 8(2): 205-215. DOI:10.1016/S0955-0674(96)80067-6 |
Tuskan G A, DiFazio S, Jansson S, et al. 2006. The genome of black cottonwood, Populus trichocarpa (Torr. & Gray)[J]. Science, 313(15): 1596-1604. |
van Wees S C, de Swart E A, van Pelt J A, et al. 2000. Enhancement of induced disease resistance by simultaneous activation of salicylate and jasmonate dependent defense pathways in Arabidopsis thaliana[J]. P Natl Acad Sci USA, 97(15): 8711-8716. DOI:10.1073/pnas.130425197 |
Voorrips R E. 2002. MapChart:Software for the graphical presentation of linkage maps and QTLs[J]. J Hered, 93(1): 77-78. DOI:10.1093/jhered/93.1.77 |
Yang T, Poovaiah B W. 2002. A calmodulin-binding /CGCG box DNA-binding protein family involve in multiple signaling pathways in plants[J]. J Biol Chem, 277(47): 45049-45058. DOI:10.1074/jbc.M207941200 |
Yin T M, DiFazio S P, Gunter L E. 2004. Genetic and physical mapping of Melampsora rust resistance genes in Populus and characterization of linkage disequilibrium and flanking genomic sequence[J]. New Phytol, 164: 95-105. DOI:10.1111/j.1469-8137.2004.01161.x |
Yin T M, Difazio S P, Gunter L E, et al. 2008. Genome structure and emerging evidence of an incipient sex chromosome in Populus[J]. Genome Research, 18: 422-430. DOI:10.1101/gr.7076308 |
Zeng Yanru(曾燕如), Huang Minren(黄敏仁), Wang Mingxiu(王明庥).2004. Construction of cDNA libraries with leaves of clones susceptible and resistant to black spot disease in poplar. Journal of Nanjing Forestry University (南京林业大学学报), 28(3):83-85.
|
Zhang J, Steenackers M, Storme V, et al. 2001. Fine mapping and identification of nucleotide binding site/leucine-rich repeat sequences at the MER locus in Populus deltoides S9-2[J]. Phytopathology, 91(1): 1069-1073. |
Zhang L, Lu Y T. 2003. Calmodulin-binding protein kinases in plants[J]. Trends Plant Sci, 8(3): 123-127. DOI:10.1016/S1360-1385(03)00013-X |