 2010, Vol. 46
2010, Vol. 46文章信息
- 杜庆章, 王博文, 王保垒, 张曼, 李百炼, 张志毅, 张德强
- Du Qingzhang, Wang Bowen, Wang Baolei, Zhang Man, Li Bailian, Zhang Zhiyi, Zhang Deqiang
- 毛白杨木材形成功能基因内SSR标记的开发及评价
- Development and Evaluation of Simple Sequence Repeat (SSR) Loci from Functional Genes Involved in Wood Formation in Populus tomentosa
- 林业科学, 2010, 46(11): 8-15.
- Scientia Silvae Sinicae, 2010, 46(11): 8-15.
-
文章历史
- 收稿日期:2010-07-08
- 修回日期:2010-08-03
-
作者相关文章



2. 北京林业大学林木花卉遗传育种教育部重点实验室 北京 100083;
3. 西北农林科技大学 杨凌 712100
2. Key Laboratory of Genetics and Breeding in Forest Trees and Ornamental Plants of Ministry of Education, Beijing Forestry University Beijing 100083;
3. Northwest A & F University Yangling 712100
为应对人类面临的全球性能源危机和温室效应的日益加剧,新能源的研发已成为世界各国优先发展的战略,纤维质生物量是最丰富的可再生资源,通过生物转化成乙醇可替代石油作液体燃料,有望解决出现的能源短缺问题(Demirbas,2005)。而森林作为最重要的陆地生态系统,为人类生存提供了约80%的纤维质生物量(Demirbas,2005)。其中,由于杨树(Populus)有生长迅速、轮伐期短、种间杂交与无性繁殖比较容易和便于遗传操作等优点,在北半球被认为是最具前途的纤维质能源树种(Li et al., 2008)。因此,深入研究木材生物合成途径,阐明木材纤维性状形成的分子机制是加速木材品质遗传改良的基础。
而目前在分子遗传学方面的主要进展是通过分子标记辅助选择(marker assisted selection,MAS)策略将影响木材纤维性状的多个基因剖分开来,可以像研究质量性状基因位点一样,对这些数量性状基因位点进行研究,研究这些基因在染色体上的位置,并寻找与其紧密连锁的分子标记,以此来对林木进行遗传改良(Brown et al., 2003; Zhang et al., 2006)。在常见的分子标记技术中,简单序列重复(simple sequence repeats,SSRs)也称微卫星DNA,由于具有多态性高、共显性遗传、检测容易且结果稳定、重复性强等优点,被认为是一种理想的分子标记(Tuskan et al., 2004)。但,先前开发的SSR位点多来自基因组的随机区域,而这些SSR多呈中性进化,与物种表型性状的形成连锁不太紧密(Tuskan et al.,2004)。与此不同,基因内的SSR由于其功能重要性,在进化过程中,一般会受到较强烈的选择压,造成与物种的表型性状紧密连锁(Li et al., 2004)。因此,开发木材形成功能基因内SSR位点,是利用该标记技术进行重要纤维品质性状分子标记辅助育种的前提。
为此,本研究以最大程度地反映毛白杨(Populus tomentosa)自然分布区域的40个基因型个体为材料,在测定木材形成相关基因序列的基础上,发现基因内不同区域SSR位点,并依据SSR位点两侧的保守区域设计SSR扩增引物。在此基础上,以杨属内白杨派(Leuce)、黑杨派(Aigeiros)、青杨派(Tacamahaca)、胡杨派(Turanga) 15个基因型个体为试验材料,对开发的20对SSR引物进行了多态性评价,证明了其在杨属内的有效性。研究结果为利用功能基因内SSR位点进行QTL分析及连锁不平衡(linkage disequilibrium,LD)作图提供了重要的科学理论依据,具有重要的应用价值。
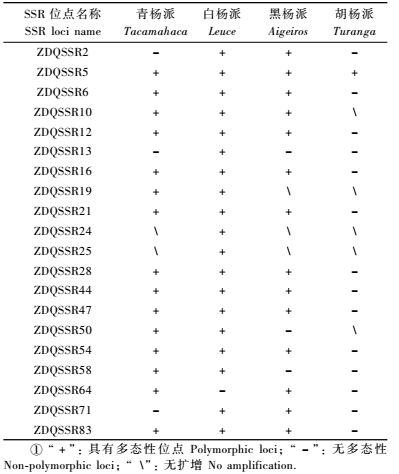
1 材料与方法 1.1 植物材料用于毛白杨木材形成相关基因内SSR发现的材料取自全国毛白杨基因库中能够最大程度地反映毛白杨天然分布范围的40株基因型个体(徐煲铧等,2009); 而用于SSR引物有效性评价的材料为表 1中杨属内15个基因型个体。
|
|

总DNA提取按DNeasy Plant Mini Kits(Qiagen,Inc.,Valencia,CA,USA)描述的方法进行。
1.3 SSR位点发现及引物开发作者所在课题组在进行杨树主干维管组织如韧皮部、形成层、未成熟木质部及成熟木质部基因表达谱芯片分析时发现许多高丰度差异表达的基因,本研究从中选取了在形成层及成熟木质部表达量超过其他组织5倍以上的16个基因作为候选序列,并设计了基因特异的PCR扩增引物。在此基础上,以1.1中描述的40株毛白杨基因型个体为材料进行PCR扩增。将PCR扩增产物进行琼脂糖凝胶电泳分离,回收、纯化目的片段后与PGEM-T载体连接,转化后挑取阳性单克隆进行序列测定,然后将每一基因片段的核苷酸序列拼接成完整的基因序列。利用MEGA4.50.4软件对每一基因的40个序列进行比对分析,从而发现基因内(启动子、5′ UTR、外显子、内含子、3′ UTR)不同区域SSR位点,并依据SSR位点两侧的保守区域设计SSR扩增引物。
1.4 SSR位点扩增及电泳检测根据设计的核苷酸引物对应用25 μL DNA聚合酶链式反应(PCR)体系,以杨属不同种及毛白杨种内不同个体提取的DNA为模板,加入2.5 μL10 × buffer,1.8 μL 25 mmol·L-1 MgCl2,1 μL10 mmol·L-1 dNTP,Taq DNA聚合酶1.0 U(以上试剂购自Promega公司),100 nmol·L-1正向和反向引物各1 μL,加适量双蒸水至25 μL。于94 ℃,3 min→(93℃,30 s →56 ℃,30 s→ 72℃,40 s),35个循环→72 ℃,5 min热循环条件下,扩增出预期长度的目的片段。扩增产物加入10 μL Loading buffer(98%甲酰胺,10 mmol·L-1 EDTA,0.25%二甲苯青,0.25%溴芬蓝),经变性后在6%的变性聚丙烯酰胺胶上电泳分离,条件是95 W,2 000 V,恒功率电泳约70 min。电泳后采用Tixier等(1997)的银染检测方法进行显色观察,记录扩增结果。
1.5 SSR引物的有效性评价利用开发的SSR引物,检测其在杨属内不同个体间的有效性扩增,并分析其多态性; 根据电泳结果,统计扩增谱带,建立0,1矩阵; 在NTSYpc2.10软件中计算种间个体的遗传相似系数(geneticsimilarity,GS)及遗传距离(genetic distance,GD),并进行个体之间的UPGMA聚类分析,最终在分子水平上确定杨属内各派间亲缘关系。
2 结果与分析 2.1 毛白杨木材形成相关基因内SSR位点发现为了检测杨树木材形成相关基因内的SSR位点,选取了在杨树主干维管组织差异表达的16个候选基因作为目标,对这些基因的5′ UTR、外显子、内含子、3′ UTR及部分基因的启动子区域进行克隆(表 2)。由表 2可见,分析的这16个基因长度为692 ~ 8 434 bp,共覆盖杨树基因组长度为79 354 bp。通过对在木材主干维管组织差异表达的16个基因在40个毛白杨基因型个体中的序列比较分析,共检测到20个SSR位点(表 3)。由表 3可见,开发的这20个SSR位点,均源于基因的非编码区,即基因的启动子、5′ UTR、内含子及3′ UTR区域。其中,有10个位点源于内含子区域,占总位点数的50.0%;而有6个则位于启动子区域(表 3)。在这20个SSR位点中,碱基重复呈现出二碱基、三碱基、五碱基、六碱基与七碱基形式,其中以二碱基重复的位点较多,占总数的55%,且尤以(AT)n,(CT)n,(TC)n较普遍(表 3)。
|
|

|
|

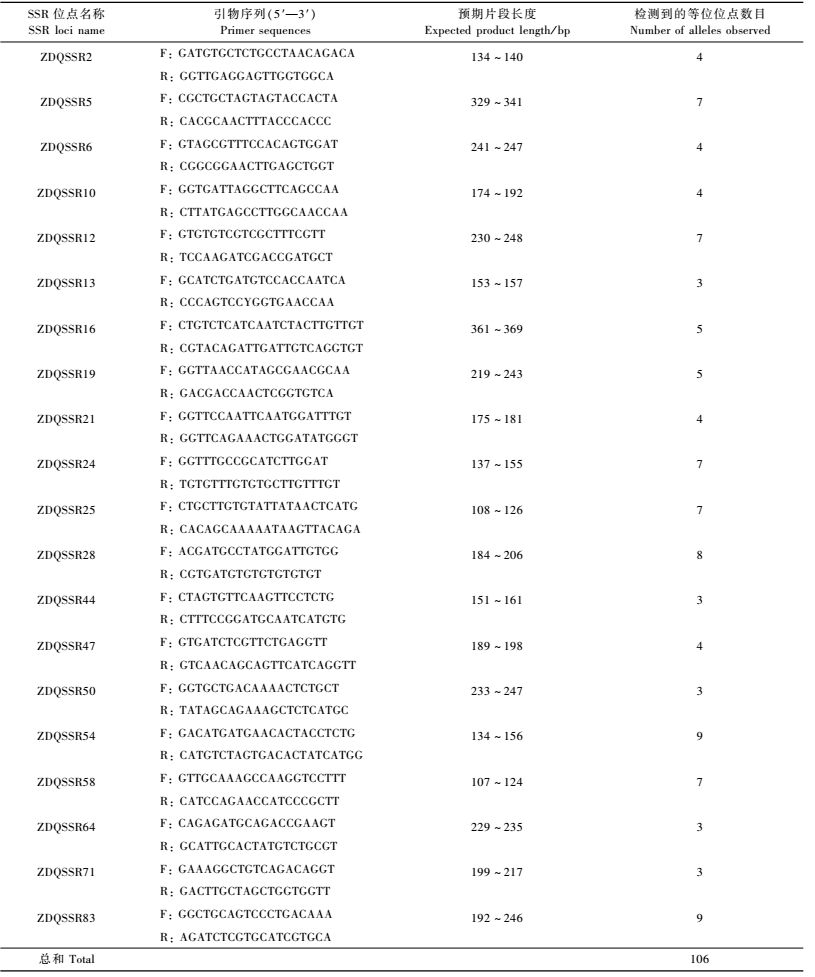
在SSR位点发现的基础上,依据其两端的保守序列,设计了20对SSR位点PCR扩增引物(表 4)。为了检测所设计的这20对引物组合在杨属内PCR扩增的有效性,以表 1所列的杨属白杨派、黑杨派、青杨派及胡杨派内15个基因型个体为材料进行PCR扩增及等位位点多样性检测,结果列于表 4与表 5。可见,除引物对ZDQSSR24与ZDQSSR24仅能在白杨派树种中进行有效扩增外,其他18对引物组合都至少能在2个派内个体中进行有效扩增,因此,在开发的SSR标记引物中,100%能够在同一派内不同树种中得到有效扩增,90%的标记可在同一属内不同种间进行有效扩增,而75%以上的标记能够延伸到同一科的柳属树种中得到扩增(表 5)。每对引物组合可检测到SSR多样性位点数为3 ~ 9个,共检测到106个多样性位点,平均5.3个。
|
|
|
|
为了检测木材形成相关基因内开发的SSR位点在毛白杨及杨属内不同种间的等位变异,比较了来源于基因内不同区域SSR位点的多样性变异。结果表明,源于基因启动子区域的SSR位点无论在毛白杨种内不同个体间还是在杨属内不同派间均显示出较高的多样性等位变异。图 1显示了在毛白杨纤维素合酶PtCesA4与PtCesA8基因启动子区域检测到的AT与AG作为重复单元的SSR位点多样性。在这2个基因的启动子区域,AT与AG单元的重复次数分别为8 ~ 34次及8 ~ 16次,显示了启动子区域SSR位点在毛白杨种内具有广泛的遗传变异; 而在杨属内不同种间也展示了较为广泛的等位位点多样性,变异范围为5 ~ 9个,平均为7.3个(表 4)。与源于启动子区域的SSR相比,位于5′ UTR的位点则较为保守,变异幅度较低,如源于PtCesA3基因5′ UTR以CT为重复单元的SSR位点等位变异程度较低,重复单元次数仅为5 ~ 7次(图 2)。同样,位于5′ UTR的SSR位点在种间检测到的等位变异多样性也较低,变异范围为3 ~ 4个,平均为3.3个(表 4)。

|
图 1 毛白杨不同个体间纤维素合酶基因启动子区域SSR位点多样性 Figure 1 SSR diversity within promoter region of PtCesA genes from different individuals in P.tomentosa A:毛白杨PtCesA4 CesA4 of P.tomentosa; B:毛白杨PtCesA8 CesA 8 of P.tomentosa. |

|
图 2 毛白杨不同个体间PtCesA3基因5′ UTR区域SSR位点多样性 Figure 2 SSR diversity within 5′ UTR region of PtCesA3 from different individuals in P.tomentosa |
基因内的SSR由于其功能重要性,在进化过程中,一般会受到较强烈的选择压,造成与物种的表型性状紧密连锁。因此,很有必要尝试开发这些基因内SSR位点在杨属不同派间分类中的应用。为此,随机选取了其中的10个SSR位点来检测杨属内不同种间的遗传进化关系。通过对SSR电泳结果显示,15份供试材料在SSR水平上表现出明显的多态性差异,且可很容易区分出不同派间的杨树个体。如选用ZDQSSR19引物组合对供试材料进行的谱带检测可明显区分出这15个杨树个体,可将其分出4类,即青杨派(编号1 ~ 4)、白杨派(编号5 ~ 10)、黑杨派(编号11 ~ 13)及胡杨派树种(编号14 ~15) (图 3)。在此基础上,计算了个体间的遗传距离及遗传相似系数。杨属各派间遗传指数变化范围较大,遗传相似系数为0.606 3 ~ 0.960 8,平均为0.783 4;遗传距离为0.100 7 ~ 0.992 5,平均为0.550 7。根据遗传距离矩阵,利用NTSYpc2.10软件进行15个基因型个体的UPGMA聚类分析(图 4),聚类结果与电泳谱带显示的结果及常规分类一致,即各派在杨属中是明显独立的,黑杨派、青杨派及胡杨派亲缘关系较近,与白杨派亲缘关系较远。

|
图 3 ZDQSSR19引物组合对15份杨属种间材料的SSR位点 Figure 3 The amplified SSR loci of 15 inter-specific individuals under Populus by ZDQSSR19 M:标准DNA分子量(pUC19 /MspⅠ) Standard DNA ladder (pUC19 /MspⅠ); 1:大青杨P.ussuriensis; 2:小叶杨P.simonii; 3:香杨P.suaveolens; 4:滇杨P.yunnanensis; 5:毛白杨P.tomentosa; 6:欧洲山杨P.tremula; 7:山杨P.davidiana; 8:银白杨P.alba; 9:新疆杨P.bolleana; 10:河北杨P.hopeiensis; 11:美洲黑杨P.deltoides; 12:箭杆杨P.nigra var.thevestina; 13:钻天杨P.nigra var.italica; 14:胡杨♀P.euphratica ♀; 15:胡杨♂ P.euphratica ♂. |

|
图 4 基于SSR标记的杨属15个不同种间个体的聚类分析 Figure 4 Cluster analysis of 15 inter-specific individuals under Populus using SSR markers |
SSR作为一种分子标记技术,与其他标记如RFLP、RAPD及AFLP等相比,具有多态性高、共显性、DNA用量少、试验重复性好、结果可靠等优点,现已被广泛应用于林木树种的遗传研究及分子标记辅助早期选择育种(Tuskan et al., 2004)。因此,开发树种特异的SSR标记是进行该物种分子遗传学及功能基因组学研究的基础,如利用SSR标记进行重要树种遗传多样性评价与核心种质构建、绘制指纹图谱与种质鉴定、重要经济性状的基因定位与分子标记辅助选择。先前虽利用基因组插入文库筛选法、微卫星富集法及数据库生物信息学搜索法开发了一批SSR标记(Yasodha et al., 2008; Yin et al., 2009),但由于开发的这些SSR位点多来自基因组的随机区域,在应用SSR标记辅助选择育种实践中则经常会出现两方面的问题:一是随机区域特别是基因间隔区虽可检测到较多的SSR位点,但由于这些区域的保守性较低,造成在一个物种中开发的SSR标记很难延伸应用到近缘物种的遗传学研究中; 另一问题是这些SSR多呈中性进化,与物种表型性状的形成连锁不太紧密,很难检测到与表型性状显著连锁的SSR位点(Vasemägi et al., 2005)。而功能基因在进化过程中,一般会受到强烈的自然选择,由于源于基因内的SSR座位与基因一样也受到了强烈的选择压,结果造成基因内的SSR多态性位点经常会与物种的表型性状的形成紧密连锁(Li et al., 2004)。若SSRs发生在基因的编码区,就可能导致产生新的蛋白质和mRNA的结构折叠,造成质量上的不同; 若位于调控区或内含子区,就可能影响基因表达水平、RNA的稳定性而导致数量上的变化(Li et al., 2004)。因此,开发功能基因内SSR位点并分析其多态性,是利用该标记技术进行重要经济性状分子标记辅助育种的前提。为此,检测木材形成功能基因内的SSR多态性位点,开发与木材纤维性状显著连锁的SSR标记是加速杨树生物质能源树种改良及纸浆材林建设的分子遗传学基础。
围绕上述目的,本文首次将在杨树主干维管组织差异表达的16个ESTs作为候选基因,直接分析这些基因的启动子、5′ UTR、外显子、内含子及3′ UTR区域在毛白杨自然群体中的多态性来开发树木功能基因内的SSR标记。通过该策略分析了覆盖杨树基因组长度为79 354 bp的基因序列,检测到了20个多态性SSR位点(表 3)。与先前在青杨派树种毛果杨(P.trichocarpa)中开发的SSR标记(Tuskan et al., 2004; Yin et al., 2009)相比,具有明显的优点:一是本文报道的是多态性的SSR位点,而在毛果杨中是基于1个基因型个体检测到具有简单序列重复的SSR位点,在不同基因型个体中不一定具有多态性(Tuskan et al., 2004; Yin et al., 2009)。二是基因内开发的SSR标记能够扩展应用到同一属内不同亚属甚至同一科内。本文开发的SSR标记中,100%能够在同一派内不同树种中有效扩增,90%的标记能够在同一属内不同种间进行有效扩增,而75%以上的标记能够延伸到同一科的柳属树种中得到扩增; 而在毛果杨中开发的SSR标记能够在美洲山杨(P.tremuloides)中得到扩增的不足80% (Yin et al., 2009)。由于基因内开发的SSR标记能够在同一属内不同种间得到有效扩增,因此应用基因内的SSR标记位点可在分子水平上将杨属4个派内的个体明显区分,分析不同派间及派内个体的遗传距离(图 4)。虽然目前基于表达序列标签(expressed sequence tag,EST)开发的SSR标记在群体遗传学及标记辅助选择育种研究中也显示了强有力的生命力,但也存在明显的不足之处,如真核生物基因内一般会存在一个至多个内含子,基于EST开发的引物对经常会落在内含子与外显子的剪切位点而不能得到扩增,在数据分析时被误认为是零等位(null allele),结果造成错误的结论(Ellis et al., 2007)。因此,究竟选用具有何种特点的SSR标记,则需要依据研究项目的需求,选用合适的SSR标记位点。如若要进行群体遗传结构及父本分析,建议选用源于基因组随机区域的SSR位点; 若要进行分子标记辅助选择育种则选用基于基因内的SSR标记较为理想。
徐煲铧, 杨晓慧, 李百炼, 等. 2009. 毛白杨纤维素合酶基因PtCesA4的克隆、表达及单核苷酸多态性分析[J]. 林业科学, 45(5): 1-10. DOI:10.11707/j.1001-7488.20090501 |
Brown G R, Bassoni D L, Gill G P, et al. 2003. Identification of quantitative trait loci influencing wood property traits in loblolly pine (Pinus taeda L.) Ⅲ. QTL verification and candidate gene mapping[J]. Genetics, 164(4): 1537-1546. |
Demirbas A. 2005. Bioethanol from cellulosic materials: a renewable motor fuel from biomass[J]. Energy Sources, 27(4): 327-337. DOI:10.1080/00908310390266643 |
Ellis J R, Burke J M. 2007. EST-SSRs as a resource for population genetic analyses[J]. Heredity, 99(2): 125-132. DOI:10.1038/sj.hdy.6801001 |
Li X, Weng J K, Chapple Clint. 2008. Improvement of biomass through lignin modification[J]. The Plant Journal, 54(4): 569-581. DOI:10.1111/j.1365-313X.2008.03457.x |
Li Y C, Korol A B, Fahima T. 2004. Microsatellites within genes: structure, function, and evolution[J]. Mol Biol Evol, 21(6): 991-1007. DOI:10.1093/molbev/msh073 |
Tixier M H, Sourdille P, Leroy P, et al. 1997. Detection of wheat microsatellites using a non-radioactive silvernitrate staining method[J]. J Genet Breed, 51: 175-177. |
Tuskan G A, Gunter L E, Yang Z K. 2004. Characterization of microsatellites revealed by genomic sequencing of Populus trichocarpa[J]. Can J For Res, 34(1): 85-93. DOI:10.1139/x03-283 |
Vasemägi A, Nilsson J, Primmer C R. 2005. Expressed sequence taglinked microsatellites as a source of gene-associated polymorphisms for detecting signatures of divergent selection in atlantic salmon (Salmo salar L.)[J]. Mol Biol Evol, 22(4): 1067-1076. DOI:10.1093/molbev/msi093 |
Yasodha R, Sumathi R, Chezhian P, et al. 2008. Eucalyptus microsatellites mined in silico: survey and evaluation[J]. J Genet, 87(1): 21-25. DOI:10.1007/s12041-008-0003-9 |
Yin Tongming, Zhang Xinye, Gunter L E, et al. 2009. Microsatellite primer resource for Populus developed from the mapped sequence scaffolds of the Nisqually-1 genome[J]. New Phytologist, 181(2): 498-503. DOI:10.1111/nph.2009.181.issue-2 |
Zhang Deqiang, Zhang Zhiyi, Yang Kai. 2006. QTL analysis of growth and wood chemical content traits in an interspecific backcross family of white poplar (Populus tomentosa × P. bolleana) × P. tomentosa[J]. Can J For Res, 36(8): 2015-2023. DOI:10.1139/x06-103 |