 2009, Vol. 45
2009, Vol. 45文章信息
- 张新叶, 宋丛文, 张亚东, 杨彦伶, 黄敏仁.
- Zhang Xinye, Song Congwen, Zhang Yadong, Yang Yanling, Huang Minren
- 杨树EST-SSR标记的开发
- Development of EST-SSR in Populus deltoides and P. euramericana
- 林业科学, 2009, 45(9): 53-59.
- Scientia Silvae Sinicae, 2009, 45(9): 53-59.
-
文章历史
- 收稿日期:2008-07-03
-
作者相关文章



2. 南京林业大学林木遗传和基因工程重点实验室 南京 210037
2. Key Laboratory of Forest Genetics and Gene Engineering, Nanjing Forestry University Nanjing 210037
SSR(simple sequence repeat,简单重复序列),又称微卫星(microsatellite),是真核生物基因组中1~6个核苷酸的串联重复,广泛分布于人类和其他动植物基因组中。SSR标记因其在动植物基因组中随机分布,呈共显性遗传,并且具有多态性高、稳定性好和操作简单等优点,在遗传和物理图谱的构建、基因定位、遗传多样性和物种进化分析、亲缘关系鉴定、DNA指纹图谱构建、比较基因组学、分子标记辅助选择育种等方面有广泛的应用前景。
近年来, 随着EST (expressed sequence tags)计划在不同物种间的扩展和研究内容的深入, 来源于不同物种、不同组织、不同细胞类型和不同发育阶段的基因表达序列的数目在公共数据库中急剧上升(http://www.ncbi.nlm.nih.gov)。这些表达序列不仅在新基因的挖掘中发挥了重要作用,而且成为寻找多态性新型分子标记的重要数据来源。目前,EST数据已被广泛用于产生EST-SSR (Berube et al., 2007; Poncet et al., 2006; Nicot et al., 2004; Scott et al., 2000)及EST-SNPs (Picoult-Newberg et al., 1999; Buetow et al., 1999)等分子标记。ESTs为SSR标记的开发提供一个巨大的、有价值的来源,能够克服传统SSR引物在开发时费用高的问题。这种标记为了与传统的SSR标记相区别,一般称之为EST-SSR标记。EST-SSR标记与基因组SSR标记相比,具有不同物种间通用性较好、开发方法简单及成本低廉等优点, 而且EST-SSR标记来自于基因的编码序列,更容易获得基因表达的信息,为功能基因的直接鉴定提供了可能性。目前EST-SSR标记已成为重要的农艺性状定位、遗传作图、遗传多样性、比较基因组学研究的新型重要工具, 如Dracatos等(2006)对黑麦草(Puccinia coronate f.sp. lolii)的冠状锈病病原菌进行EST-SSR标记开发和相关研究,Chen等(2008)利用EST-SSR标记构建柑桔(Citrus sinensis)和枳(Poncirus trifoliata)的遗传图谱,Choudhary等(2008)从鹰嘴豆(Cicer arietinum)中开发出EST-SSR标记并应用于其相关物种的等位基因变异分析,Simko(2009)从莴苣(Lactuca sativa)中开发EST-SSR并应用于群体遗传结构分析,Varshney等(2005b)利用来自大麦(Hordeum vulgare)的EST-SSR标记对小麦(Triticum aestivum)、黑麦(Secale cereale)和水稻(Oryza sativa)进行种间通用性及比较作图分析等。
本研究利用自行测序产生的2万余条杨树EST序列,在EST-SSR标记开发方面进行初步探讨,旨在评价杨树EST-SSR标记开发的可行性,并通过具体的相关分析,了解杨树EST-SSR的基本特性,为这些标记应用于其他相关研究奠定基础。
1 材料与方法 1.1 EST-SSR发现 1.1.1 EST序列来源及拼接所采用的EST序列在GenBank中的序列登录号为CX167465-CX187487。这些EST序列的测定主要是为了研究杨树在黑斑病致病过程中的基因表达谱情况。以美洲黑杨Ⅰ-69[Populus deltoides ‘Lux’ (Ⅰ-69/55)]及欧美杨Ⅰ-45(P. euramericana ‘Ⅰ-45’)经杨树黑斑病致病菌Marssonina brunnea f. sp. brunnea接种72 h后构建的2个cDNA文库为材料,从中随机挑取克隆进行5′测序,共获得21 657条EST序列,通过去载体序列、去低质量序列等处理后,共获得有效长度大于100 bp的序列20 023条。为了充分挖掘和利用这些EST序列信息,本研究即利用这20 023条EST序列进行相关EST-SSR标记的开发研究。
1.1.2 EST-SSR标记的发现首先利用Phrap软件对这20 023条EST序列进行重叠群的组装拼接, 然后利用软件SSRIT (simple sequence repeat identification tool)在线(http://www.gramene.org/de/seaches/ssrtool)对拼接过的非重复UniGene序列进行SSR序列查找。查找标准为:重复次数不小于5次的二核苷酸、三核苷酸、四核苷酸和五核苷酸重复序列。然后再应用引物设计软件Primer 3.0(http://fokker.wi.mit.edu/cgi-bin/primer3/primer3-www.cgi)对含有SSR的UniGene序列进行引物设计。SSR引物设计原则为:EST序列长度大于100 bp;引物长度为18~25 bp;退火温度Tm值55~60 ℃;GC含量40%~60%;PCR扩增产物长度为100~300 bp。
1.2 EST-SSR结果验证以我国湖北省内种植较多的6个杨树品种Ⅰ-63杨[Populus deltoides ‘Lux’ (Ⅰ-63/51)], Ⅰ-69杨[Populus deltoides ‘Lux’ (Ⅰ-69/55)], Ⅰ-72杨(Populus deltoides ‘Ⅰ-72’),南林-95杨(Populus×euramericana ‘NL95’),南林-895杨(Populus×euramericana ‘NL895’),楚林-1(Populus×euramericana ‘CL1’)为植物材料, 各选取嫩叶数片, 利用改进的CTAB法进行基因组总DNA提取。PCR反应体系15 μL:10 μmol·L-1Tris-HCl (pH 8.3), 50 mmol·L-1 KCl, 2.0 mmol·L-1 MgCl2,0.01% gelatin, 0.1 mg BSA, 200 μmol·L-1 dNTP(Promega),引物2.0 μmol·L-1, Taq聚合酶(Takara)0.5 U,10 ng基因组DNA。PCR反应在仪器GenAMP 9700(ABI)上进行,反应程序采用Touch-down PCR:94 ℃ 5 min; 94 ℃ 30 s, 59 ℃ 30 s(Δ℃=-1.0 ℃),72 ℃ 30 s, 9个循环;94 ℃ 30 s,55 ℃ 30 s,72 ℃ 30 s, 21个循环;72 ℃ 3 min; 4 ℃保存。
采用荧光PCR产物的检测方法。引物合成时用荧光FAM标记5′端寡核苷酸引物(由大连宝生物公司合成)。PCR产物稀释50~100倍后加2 μL于8 μL上样缓冲液[HiDi formamide, ROX400 standard(ABI)]中,然后95 ℃变性5 min,迅速置于冰水上冷却。在ABI 3100遗传分析仪上电泳并收集数据。软件Genemapper 3.7(ABI)用于原始信号收集后的片断大小读取及位点判读。
2 结果与分析 2.1 EST序列拼接及EST-SSR标记分析利用软件Phrap共对20 023条有效长度大于100 bp的EST序列进行拼接分析。共获得10 816个非重复序列(UniGene), 其中包括3 734个重叠群(Contig), 7 082个独立的ESTs(Singletons)。UniGene序列长度主要集中在500~800 bp。在3 734个Contigs序列中, Contig size (组成Contig的EST序列数)最大值为92个EST序列, 平均值为3.45个EST序列。碱基最长的Contig序列长度为3 499 bp,由34条EST序列组成。
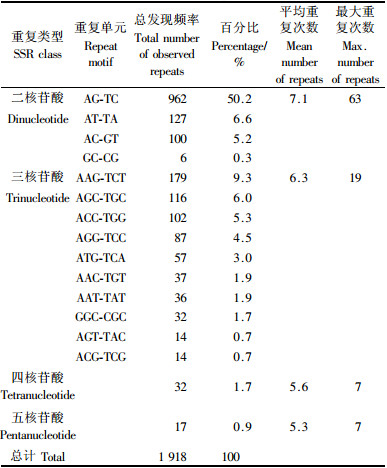
为了减少所发现SSR的冗余度, 从拼接过的10 816个UniGene序列中进行SSR分析,结果共发现1 604个UniGene序列中含有SSR序列,占总UniGene序列数的14.8%。由于部分序列含有1个以上的SSR重复序列,共在1 604个UniGene序列中发现1 918个SSR序列,其中,有231个UniGene序列含2个SSR重复序列,32个UniGene序列含3个SSR重复序列,5个UniGene序列含4个SSR重复序列,1个UniGene序列含5个SSR重复序列,其他UniGene序列只含1个SSR重复序列。在这1 604个UniGene序列中,序列最长的是Contig6311,长度为3 499 bp,但只含有1个SSR序列,而含有最多SSR序列(5个)的UniGene序列Contig4912,长度仅为705 bp。
由于所发现的重复序列单元复杂多样,为分析方便,根据Jurke等(1995)的分类标准,将重复单元分成16类,其中二核苷酸重复4类,分别为(AT)n-(TA)n, (AG)n-(TC)n, (AC)n-(GT)n和(GC)n-(CG)n; 三核苷酸重复10类,分别为(AAT)n-(TAT)n, (AAG)n-(TCT)n, (AAC)n-(TGT)n, (ATG)n-(TCA)n, (AGT)n-(TAC)n, (AGG)n-(TCC)n, (AGC)n-(TGC)n, (ACG)n-(TCG)n, (ACC)n-(TGG)n和(GGC)n-(CGC)n。由于四核苷酸重复和五核苷酸重复相对较少, 所以不对四核苷酸重复和五核苷酸重复进行具体分类,只将其分别归为一类。从表 1中可以看出这16类核苷酸重复各自所出现的频率,二核苷酸重复占总SSR的62.3%,三核苷酸重复占35%, 四核苷酸和五核苷酸共占2.6%。其中, 在二核苷酸重复中,(AG)n所占比例最高,达50%以上;在三核苷酸重复中,(AAG)n所占比例最高。这与Tuskan等(2004)从基因组序列中发现的各类SSR重复所占比例结果存在较大差异,他们发现最丰富的二核苷酸重复为(AT)n, 最丰富的三核苷酸重复为(AAT)n。在本研究所发现的1 918个SSR重复序列中,最长的二核苷酸重复单元为[GA]63,最长的三核苷酸重复单元为[GCA]19,最长的四核苷酸重复单元有5个:[ACAT]7,[CATA]7,[CCAT]7,[CCTC]7及[CTCC]7;最长的五核苷酸重复单元为[CAATC]7。
从表 1中还可以看出,所观察到的重复单元的重复次数平均值随重复单元中核苷酸数量的增加而减少。这与其他物种研究中观察到的结果相一致。另外,不同SSR重复单元的出现频率与其重复次数的相关性统计分析结果(图 1)表明:随着重复次数(或SSR长度)的增加,所有二核苷酸、三核苷酸、四核苷酸及五核苷酸SSR序列的频率均呈明显下降趋势,且当SSR的长度达到30 bp以上时(二核苷酸SSR的主要重复次数为5~15次,三核苷酸SSR的主要重复次数为5~12次,四、五核苷酸SSR的主要重复次数均为5~7次),这些不同类型的SSR序列出现的频率都几乎接近零。
|
|

|
图 1 SSR重复类型与其重复次数的关系分布图 Figure 1 Distribution of the number of repeats by repeat type |
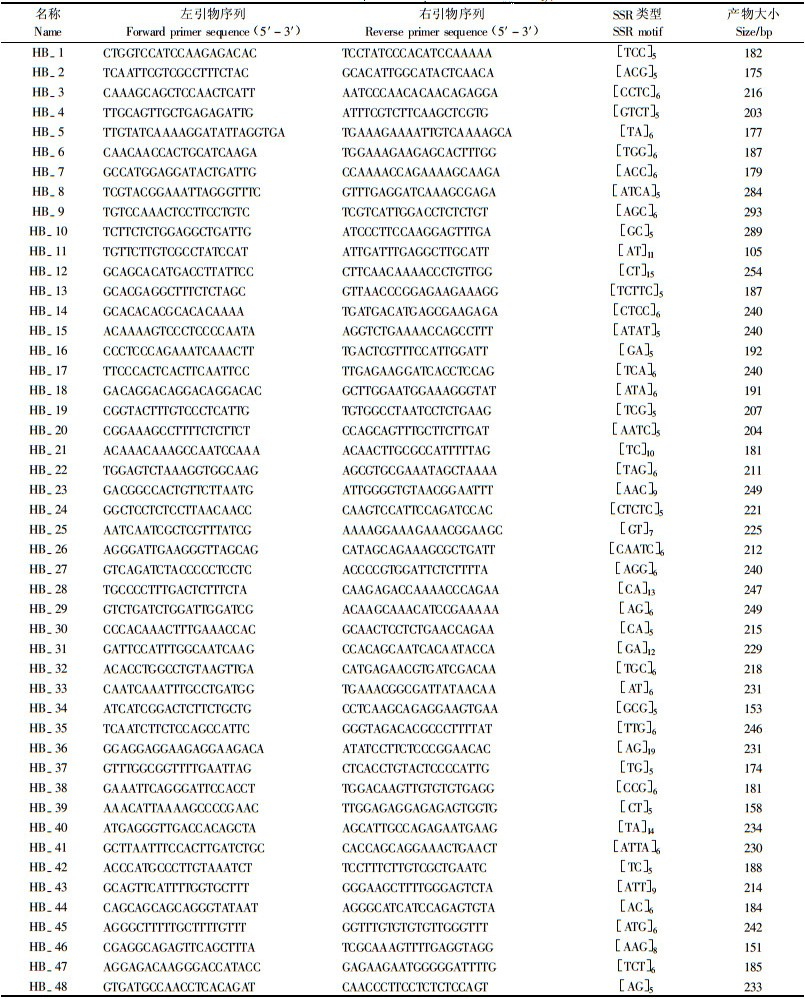
在含有SSR重复单元的非重复序列中随机抽取100条序列进行SSR引物设计, 根据设计结果, 选取48对EST-SSR引物进行生物合成, 其中分别含有二核苷酸、三核苷酸、四核苷酸及五核苷酸重复单元(表 2)。首先利用一个DNA样品(Ⅰ-69)对这48对引物进行初筛, 结果有5对引物没有扩增产物, 占总比例的10.4%;同时发现,在有产物的43对引物中有9对引物产生的扩增片段大小与预期大小不符, 其中引物HB_19(756 bp)和HB_20(650 bp)产生的片段大小远远大于预期片段(数据未列出)。然后在有产物的43对引物中随机抽取12对, 利用6个不同的杨树品种对其进行多态性检验。结果发现有7对引物在不同样品间产生多态性分离,其他5对引物在不同样品间获得的产物大小一致,没有分离。
|
|
从图 2中,可以很清楚地看到标记HB_1在6个不同杨树品种间产生的多态分离情况。本研究利用DNA测序仪及荧光标记对PCR产物进行检测, 精度可以达到1 bp, 其分析准确性明显高于琼脂糖凝胶及PAGE胶的结果。这些EST-SSR标记的验证结果发现, 相对传统开发基因组SSR而言,利用数据库中现有的大量杨树EST序列信息,开发杨树EST-SSR标记是切实可行的又一开发捷径。

|
图 2 引物HB_1在6个杨树品种中的多态性分离 Figure 2 Polymorphism in 6 Popolus individuals for primer HB_1 1.Ⅰ-63杨Populus deltoides ‘Lux’ (Ⅰ-63/51);2.Ⅰ-69杨Populus deltoides ‘Lux’ (Ⅰ-69/55);3. Ⅰ-72杨Populus deltoides ‘Ⅰ-72’; 4.南林-895杨Populus × euramericana ‘NL895’; 5.南林-95杨Populus × euramericana ‘NL95’; 6.楚林-1杨Populus × euramericana ‘CL1’. |
在本研究中发现至少含有一个二核苷酸、三核苷酸、四核苷酸或五核苷酸SSR重复单元的非重复序列有1 604条, 占非重复序列的14.8%,这远高于Kantety等(2002)从大麦、小麦、玉米(Zea mays)、高梁(Sorghum bicolor)、水稻EST数据库中开发SSR标记的平均水平3.2%,更高于Berube等(2007)从火炬松(Pinus taeda)和白云杉(Picea glauca)、北美云杉(Picea sitchensis)的EST数据库中开发EST-SSR标记的水平, 他们发现只有1.1%的EST序列中含有SSR, 也明显高于Nicot等(2004)从小麦170 746条EST中开发出3 530个SSR标记的水平。同时还发现, 在本研究中10 816个非重复序列总长度为7 450 819 bp, 平均每3.88 kb的EST序列发现1个SSR。而在火炬松和云杉中平均每49.8 kb出现1个SSR, 拟南芥(Arabidopsis thaliana)中平均每14 kb、水稻中平均每19 kb各出现1个SSR, 与这些研究结果相比,从杨树中开发EST-SSR标记的强度比其他物种要高。造成如此明显差别的原因主要有2点:一是本研究首先对所有的EST序列进行了序列拼接,20 023条有效EST序列拼接为10 816条非重复序列, 然后在非重复序列中发现SSR标记, 这一步在很大程度上提高了SSR标记发现的比例;二是在发现SSR标记时, 不同研究设置的参数存在明显不同。如Kantety等(2002)的研究对于二、三核苷酸重复单元设置最小长度为18 bp,对于四核苷酸重复单元设置最小长度为20 bp;Berube等(2007)设置二、三、四核苷酸重复单元最小长度与Kantety等(2002)的设置相同,另外设置五核苷酸重复次数最小值为5, 六核苷酸重复次数最小值为4;Nicot等(2004)的研究是设置所有基序重复序列的最小长度为12 bp;而本研究是设置所有SSR重复单元的最小重复次数为5次, 也就是说二核苷酸重复单元的最小长度为10 bp, 三核苷酸最小长度为15 bp, 四核苷酸最小长度为20 bp, 五核苷酸的最小长度为25 bp。由于二核苷酸和三核苷酸重复单元在各物种中出现的比例都是最高的, 所以在本研究中, 设置二、三核苷酸的最小长度小于其他研究, 也明显提高了SSR发现的比例。本研究在对EST-SSR结果的验证试验中,发现最小重复次数为5次的二、三核苷酸SSR标记也表现出了很好的多态性分离,说明重复次数设置为5,对杨树SSR标记开发来说,是一个比较好的且可以进一步探讨的参数。其实,从非冗余EST序列中开发SSR标记,其频率更能准确地反映出SSR在基因组相关转录部分中的密度和分布状况(Varshney et al., 2005a)。
3.2 重复单元频率由于大部分EST序列由外显子区组成,在它们被翻译成蛋白质时受到frameshift突变的选择强度较大,因为密码子以三核苷酸为一功能单位,因此由插入缺失突变造成的三核苷酸位移不会给一个表达基因的阅读框造成太大影响(Metzgar et al., 2000)。由于这个原因,三核苷酸重复预计是从EST序列中发现最多的SSR类型,这在谷类作物的相关研究中得到了验证(Kantety et al., 2002; Gao et al., 2003; Temnykh et al., 2001; Varshney et al., 2002)。但本研究的发现结果与Berube等(2007)从火炬松和云杉发现EST-SSR得到的结果一致,都是二核苷酸SSR是所发现类型中最丰富的,而非三核苷酸重复。是否这是林木树种基因组的一大特征,还需更多其他树种的相关研究证明。综合同类研究发现,在二核苷酸重复序列中,AG/TC重复出现频率最高,在Kantety等(2002)的研究中达16.5%,本试验中高达50.2%,在Temnykh等(1999)对水稻的研究中也是最丰富的。这主要是由于二核苷酸重复可以出现在依附于阅读框并翻译成不同氨基酸的多个密码子中,如AG/TC基序可以在mRNA中以GAG,AGA,UCU及CUR密码子形式出现,并各自翻译形成氨基酸Arg, Glu, Ala和Leu,而Ala和Leu又各以8%和10%的高频率出现在蛋白质中(Lewin, 1994),这可能是AG/TC重复在EST序列中出现如此高频率的一个原因。在本研究中,只发现了6个GC/CG重复, 占发现SSR总数的0.3%,该结果与Kantety等(2002)及Cho等(2000)的发现结果完全一致,GC/CG型SSR在他们的研究中也是发现最少的一类重复序列。至于三核苷酸重复中不同类型的发现频率,在不同研究中结果不同。在本研究中,类型最丰富的三核苷酸重复为AAG,占SSR发现总数的9.3%;在Nicot等(2004)对小麦EST的研究中发现(CCG)n最丰富,而Kantety等(2002)对小麦的研究却是AAC重复最丰富,在火炬松和云杉(Berube et al., 2007)中则发现是ACG类型最丰富。综合在本研究中发现的所有SSR类型,可以很清楚地看到随着SSR序列中重复次数的增加,其SSR出现的频率随之减少(图 1),这与Temnykh等(2001)在水稻中及Varshney等(2002)在一些谷物中发现的结果相一致,且下降趋势都是很突然的。这些结果比较符合Samadi等(1998)的假设,序列越长的SSR经受的选择压力越大。因为当整个SSR的长度为30~50 bp时,所有重复中的SSR分布都几乎接近零。
3.3 标记验证从本研究设计的SSR引物验证结果来看,约10%的引物没有得到扩增产物,约19%的引物得到的扩增产物大小与预期片段大小不符。与同类研究相比,无论是从基因组中开发SSR标记,还是从EST序列中开发SSR标记,本研究得到的结果都比较理想。在报道的不同研究中,成功的扩增比例一般为60%~90% (Thiel et al., 2003; Kota et al., 2001; Yu et al., 2004; Saha et al., 2004; Gupta et al., 2003; Scott et al., 2000)。造成扩增没有产物的可能原因大致包括:1)EST-SSR引物对的1端或2端正好位于1个剪切位点上; 2)引物对包含了较大的内含子, 产生的PCR产物无法在电泳图中显示出来; 3)用于设计引物的序列本身存在问题或来自比较荒诞的cDNA克隆。另外, 与基因组SSR相比, EST-SSR的扩增产物大小更易偏离预期产物大小(Thiel et al., 2003; Kota et al., 2001; Yu et al., 2004; Nicot et al., 2004), 这可能是由于:1)在对应的基因组序列中出现了小内含子或插入/缺失突变, 这在相关序列分析中得到了证实(Saha et al., 2004); 2)由于引物侧翼序列的复杂性较低, 造成引物的非特异性结合; 3)由于基因组染色体复制, EST序列的复制品分布于相同或不同的染色体; 4)EST属于多基因家族, 对应的引物设计位于其保守区内。关于EST-SSR的多态性问题, 58%的引物对在本研究利用的6个不同杨树品种中表现出多态性分离。Nicot等(2004)对小麦的研究中, 53%的引物对在8个不同栽培品种中表现出多态性, Guyomarc’h等(2002)的研究中, 有55%的引物表现多态性, 这些结果与本研究的结果基本一致。
总之, 本研究及其他同类研究的结果表明, 利用公共数据库中的EST及其他cDNA信息资源开发SSR标记已成为一项切实可行的技术。随着数据库中更多不同物种DNA序列的产生, 基于这些序列产生的相关新型标记技术会在更多领域更深层次的研究中得到更广泛的应用。
Berube Y, Zhuang J, Rungis D, et al. 2007. Characterization of EST-SSR in loblolly pine and spruce[J]. Tree Genetics & Genomes, 3: 251-259. |
Buetow K H, Edmonson M N, Cassidy A B. 1999. Reliable identification of large numbers of candidate SNPs from public EST data[J]. Nat Genet, 21(3): 323-325. DOI:10.1038/6851 |
Chen C X, Bowman K D, Choi Y A, et al. 2008. EST-SSR genetic maps for Citrus sinensis and Poncirus trifoliate[J]. Tree Genetics & Genomes, 4(1): 1-10. |
Cho Y G, Ishii T, Temnykh S, et al. 2000. Diversity of microsatellites derived from genomics libraries and GenBank sequence in rice (Oryza sativa)[J]. Theor Appl Genet, 100: 713-722. DOI:10.1007/s001220051343 |
Choudhary S, Sethy N K, Shokeen B, et al. 2009. Development of chickpea EST-SSR markers and analysis of allelic variation across related species[J]. Theor Appl Genet, 118: 591-608. DOI:10.1007/s00122-008-0923-z |
Dracatos P M, Dumsday J L, Olle R S, et al. 2006. Development and characterization of EST-SSR markers for the crown rust pathogen of ryegrass (Puccinia coronate f.sp. lolii)[J]. Genome, 49: 572-583. DOI:10.1139/g06-006 |
Gao L, Tang J, Li H, et al. 2003. Analysis of microsatellites in major crops assessed by computational and experimental approaches[J]. Mol Breed, 12: 245-261. DOI:10.1023/A:1026346121217 |
Gupta P K, Rustgi S, Sharma S, et al. 2003. Transferable EST-SSR markers for the study of polymorphism and genetic diversity in bread wheat[J]. Mol Genet Genomics, 270: 315-323. DOI:10.1007/s00438-003-0921-4 |
Guyomarc'h H, Sourdille P, Charmet G, et al. 2002. Characterisation of polymorpphic microsatellite markers from Aegilops tauschii and transferability to the D-genome of bread wheat[J]. Theor Appl Genet, 104: 1164-1172. DOI:10.1007/s00122-001-0827-7 |
Jurke J, Puthiyagoda C. 1995. Simple repeat DNA sequence from primates:compilation and analysis[J]. J Mol Evol, 40: 120-126. DOI:10.1007/BF00167107 |
Kantety R V, La Rota M, Matthews D E, et al. 2002. Data mining for simple sequence repeat in expressed sequence tags from barley, maize, rice, sorghum, and wheat[J]. Plant Mol Biol, 48: 501-510. DOI:10.1023/A:1014875206165 |
Kota R, Rajeev K V, Thiel T, et al. 2001. Generation and comparison of EST-derived SSRs and SNPs in barley (Hordeum vulgare L.)[J]. Hereditas, 135: 145-151. |
Lewin B. 1994. Genes V. New York, Oxford University Press.
|
Metzgar D, Bytof J, Wills C. 2000. Selection against framshift mutations limits microsatellite expansion in coding DNA[J]. Genome Res, 10: 72-80. |
Nicot N, Chiquet V, Gandon B, et al. 2004. Study of simple sequence repeat (SSR) markers from wheat expressed sequenced tags (ESTs)[J]. Theor Appl Genet, 109: 800-815. DOI:10.1007/s00122-004-1685-x |
Picoult-Newberg L, Ideker T E, Pohl M G, et al. 1999. Mining SNPs from EST databases[J]. Genome Res, 9: 167-174. |
Poncet V, Rondeau M, Tranchant C, et al. 2006. SSR mining in coffee tree EST databases:potential use of EST-SSRs as markers for the Coffea genus[J]. Mol Gen Genomics, 276: 436-449. DOI:10.1007/s00438-006-0153-5 |
Saha M C, Mian M A, Eujayl I, et al. 2004. Tall fescue EST-SSR markers with transferability across several grass species[J]. Theor Appl Genet, 109: 783-791. DOI:10.1007/s00122-004-1681-1 |
Samadi S, Artiguebielle E, Estoup A, et al. 1998. Density and variability of dinucleotide microsatellites in the parthenogenetic polyploid snail Melanoides tuberculata[J]. Mol Ecol, 7: 1233-1236. DOI:10.1046/j.1365-294x.1998.00405.x |
Scott K D, Eggler P, Seaton G, et al. 2000. Analysis of SSRs derived from grape ESTs[J]. Theor Appl Genet, 100: 723-726. DOI:10.1007/s001220051344 |
Simko I. 2009. Development of EST-SSR markers for the study of population structure in Lettuce (Lactuca sativa L.)[J]. Journal of Heredity, 100: 256-262. DOI:10.1093/jhered/esn072 |
Temnykh S, DeClerck G, Lukashova A, et al. 2001. Computational and experimental analysis of microsatellite in rice (Oryza sativa L.):frequency, length variation, transposon associations, and genetic marker potential[J]. Genome Res, 11: 1441-1452. DOI:10.1101/gr.184001 |
Temnykh S, Park W D, Ayers N, et al. 1999. Mapping and genome organization of microsatellites in rice (Oryza sativa)[J]. Theor Appl Genet, 100: 698-712. |
Thiel T, Michalek W, Varshney R K, et al. 2003. Exploiting EST databases for the development of cDNA derived microsatellite in barkey (Hordeum vulgare L.)[J]. Theor Appl Genet, 106: 411-422. DOI:10.1007/s00122-002-1031-0 |
Tuskan G A, Gunter L E, Yang Z K, et al. 2004. Characterization of microsatellites revealed by genomic sequencing of Populus trichocarpa[J]. Can J For Res, 34: 85-93. DOI:10.1139/x03-283 |
Varshney R K, Graner A, Sorrells M E. 2005a. Genic microsatellite markers in plants:features and applications[J]. Trends in Biotechnology, 23(1): 48-55. DOI:10.1016/j.tibtech.2004.11.005 |
Varshney R K, Sigmund R, Borner A, et al. 2005b. Interspecific transferability and comparative mapping of barley EST-SSR markers in wheat, rye and rice[J]. Plant Science, 168: 195-202. DOI:10.1016/j.plantsci.2004.08.001 |
Varshney R K, Thiel T, Stein N, et al. 2002. In silico analysis on frequency and distribution of microsatellites in ESTs of some cereal species[J]. Cell Mol Biol Lett, 7: 537-546. |
Yu J K, Dake T M, Singh S, et al. 2004. Development and mapping of EST-derived simple sequence repeat (SSR) markers for hexaploid wheat[J]. Genome, 47(5): 805-818. DOI:10.1139/g04-057 |