 2009, Vol. 45
2009, Vol. 45文章信息
- 刁开盛, 尹显洪, 王海军.
- Diao Kaisheng, Yin Xianhong, Wang Haijun
- 松香枞酸结构和性质的理论研究
- Theoretical Investigation on Structure and Property of Abietic Acids in Rosin
- 林业科学, 2009, 45(8): 117-123.
- Scientia Silvae Sinicae, 2009, 45(8): 117-123.
-
文章历史
- 收稿日期:2008-04-07
-
作者相关文章


2. 江南大学化学与材料工程学院 无锡 214122
2. School of Chemical and Material Engineering, Jiangnan University Wuxi 214122
松香的主要成分是树脂酸,其中包括枞酸、长叶松酸、新枞酸和左旋海松酸,这4种树脂酸具有相同的烷基氢化菲骨架结构和羧基官能团,差别仅在于共轭双键所处的位置不同。由于测量手段和条件的限制以及该类化合物的特殊性,长期以来人们对其结构的研究主要集中于共轭双键的定位这一层面,很少深入到具体的微观结构,因而有必要采用包括理论和试验的方法手段确定松香类化合物的分子结构。随着现代分子结构测定技术的进步,近年来,人们将现代分子结构研究方法应用于松香化学领域,采用单晶X-射线衍射分析法测定枞酸的晶体结构和分子结构(Okada et al., 1994),用气质联用测定氢化松香中性部分的化学组成(段文贵等,2002)。
计算化学方法不使用化学药剂,非常符合目前对绿色化学的要求,因此其方法在分子结构模拟、药物分子设计、分子组装和分子识别等(Kang et al., 2006;Tan et al., 2003;Legon,1995;Lenoir et al., 2003;Warren et al., 1993)的研究中发挥了很大的作用,但罕有用理论方法对松香产品的详细研究,曾有文献报道(段文贵等,2003)用量子化学的半经验算法(MNDO)对4种主要树脂酸进行几何构型优化及相对能量计算,进而分析和预测此类树脂酸的分子构型及相对稳定性。目前,精确度更好的从头算法和密度泛函法已经成为计算化学的主流,因此有必要考察新的理论方法对研究松香类产物的可行性。另一方面,基本热力学参数如自由能、焓和热容等是研究一种物质的重要参数,对了解该化合物的性质和反应性能有参考指导作用。由于松香中杂质众多,文献报道的只有松香混合物的热力学性质(宋湛谦,2002;张运明等,1995),对枞酸、长叶松酸、新枞酸和左旋海松酸这几种树脂酸的热力学参数还缺少文献值或不易查找,有必要运用包括理论计算在内的方法得到这些树脂酸各自的热力学参数。
本文对枞酸分子结构进行优化,找出最有可能的分子基态构型,并将结果与文献报道的MNDO法理论值和试验测定值进行比较,考察不同理论计算方法在研究该类化合物中的适应性;同时用DFT法(Becke,1993;Hohenberg et al., 1964;Kohn et al., 1965;Pople et al., 1992;Wong,1996)对枞酸、长叶松酸、新枞酸和左旋海松酸的分子进行几何全优化和能量分析,得到相应分子的热力学参数。本文研究表明,用计算化学方法对松香类化合物进行模拟研究是可行的,这为林产化工领域的开发研究引入了新的方法和内容,也为实验室研究松香类化合物提供理论依据,具有指导意义。
1 方法采用从头算Hartree-Fock(HF)法,在6-31G机组水平下对枞酸不同构型进行结构优化和能量分析,得出分子基态能量最低的稳定构型。然后在更高的函数机组6-31+G**(Frisch et al., 1984;Russo et al., 2003)水平下,用HF法和密度泛函(DFT)方法对该分子基态进行几何构型全优化,所有计算都通过频率分析确认所得到的分子构型为基态构型并获得零点能校正值;密度泛函(DFT)方法使用了Becke3参数交换函数和Lee Yang Parr关联函数即B3LYP方法(Becke,1993)。得到的几何参数与文献报道值进行分析比较。采用密度泛函B3LYP法在6-31+G**水平下进行相应计算获得相应分子结构的热力学参数。所有计算都采用Gaussian 03程序包完成。
2 结果与讨论 2.1 枞酸分子的稳定几何构型通过分子力学模拟筛选发现枞酸分子最有可能存在的8种构型(图 1),分别考虑甲基C19和C20的位置、C18羧基的空间取向和H27的空间取向;接着在HF/6-31G水平对这些可能的构型进行结构优化。每种构型的频率分析都没有发现虚频,从而确认所得到的都为分子基态构型。表 1列出了8种可能构型的总能量及其相对值(以能量最低者为基准),最高占有分子轨道(highest occupied molecular orbital,HOMO)和最低空分子轨道(lowest unoccupied molecular orbital,LUMO)之间的能级差——能隙(ε)。

|
图 1 可能存在的枞酸分子结构模型 Figure 1 The possible structural model of abietic acid |
|
|
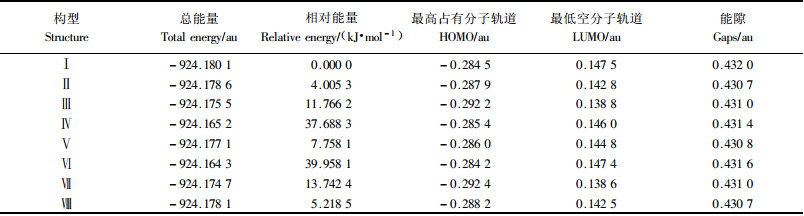
一般来说,按最低能量原理,分子总能量越低则该分子基态结构越稳定;另一方面,前线分子轨道能隙越大则受激发出现电子跃迁的几率越低,相应分子基态结构也更稳定。表 1的能量分析结果表明,构型Ⅰ具有最低的总能量,其余7种构型的总能量要比它高出4.005 3~39.958 1 kJ·mol-1不等;同时,构型Ⅰ也具有最高的前线分子轨道能隙(ε)0.432 0 au(1 au=2 625.5 kJ·mol-1)。这些都表明了枞酸分子能量最低最稳定的是构型Ⅰ(全分子构型见图 2)。

|
图 2 枞酸分子最稳定构型Ⅰ的全分子结构图 Figure 2 The whole set of most stable structure (Ⅰ) of abietic acid |
为了考察MNDO,HF和DFT方法中哪一种理论方法更适合于松香类化合物的研究,本文运用从头算Hartree-Fock(HF)和密度泛函(DFT)中的B3LYP方法,在更高函数机组6-31+G**上,重新对枞酸最稳定的分子构型Ⅰ进行几何构型全优化,得到相应的几何结构参数,并与文献报道的理论和试验数据进行比较分析。
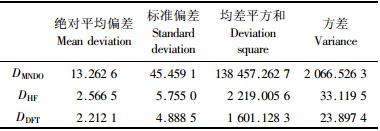
表 2给出了枞酸分子中能量最低最稳定的构型Ⅰ用3种理论方法计算得到的几何参数:半经验法MNDO得到的几何参数VMNDO(段文贵等,2003),HF的几何参数VHF和密度泛函DFT的几何参数VDFT,以及在实验室用单晶X-射线衍射分析法测得的几何结构参数VExp(Okada et al., 1994),并列出每种理论参数与单晶衍射试验测定值的相应偏差DMNDO,DHF和DDFT。从理论计算得到枞酸分子的键长、键角及二面角等几何结构参数与实测值比较来看大多数一致(表 2)。与半经验算法MNDO比较,从头算法(HF)和密度泛函法(DFT)的计算结果与实测值更加吻合,比如对二面角47# C4C3C2C1,48#C5C4C3C2,59# C14C8C7C6和61# C16C15C13C12,半经验算法MNDO理论值与试验测定值的偏差分别为-9.3°,6.6°,357.4°和102.5°,而HF法偏差分别为-1.505 17°,0.918 73°,0.494 21°和4.334 29°,密度泛函DFT的偏差最小,分别只有-1.527 09°,0.917 16°,0.289 12°和2.144 20°,显然密度泛函DFT的计算结果与单晶试验测定值更加接近。同样,在涉及分子间氢键的2个二面角参数67#O23C18C4C3和68#O24C18C4C3,理论值与真实测定值的差也是密度泛函法DFT的最小,从头算HF法其次,而半经验MNDO法与真实测定值相差最大。
|
|

表 3列出了3种理论计算方法得到的几何参数分别与单晶衍射测定试验结果之差值的误差分析。从误差分析结果来看,无论是绝对平均偏差、标准偏差、均差平方和还是方差,密度泛函法(DFT)和从头算Hartree-Fock方法(HF)都比半经验方法(MNDO)具有更高可信度,比如半经验算法的标准偏差为45.459,从头算Hartree-Fock方法(HF)的标准偏差是5.754 953,而密度泛函法(DFT)的标准偏差只有4.888 501。以上结果可知,密度泛函法(DFT)计算得到的几何参数与试验测定值的偏差最小,更适合用来对此类体系进行理论研究。
|
|
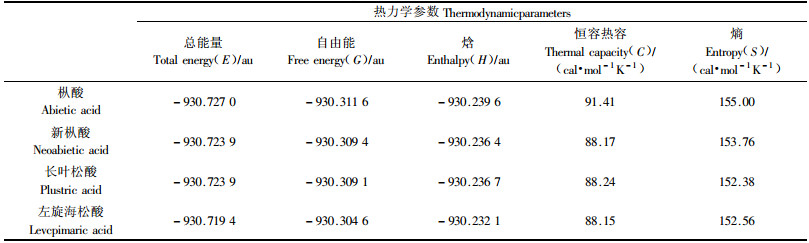
基本热力学参数如自由能、焓和热容等是研究一种物质的重要参数,对了解该化合物的性质和反应性能有参考指导作用,但由于松香酸中杂质众多,报道的都是松香混合物的热力学性质(宋湛谦,2002;张运明等,1995)。本文运用密度泛函的B3LYP的泛函在6-31+G**水平,对枞酸、长叶松酸、新枞酸和左旋海松酸这4种树脂酸分子的热力学性质进行计算,分析中使用了正则系统的理想气体标准展开,获得了298.15 K,101.325 kPa下的热力学参数。表 4列出了经过原始零点能修正后的总能量E、吉布斯自由能G、焓H、恒容热容C和熵S理论值。为便于直观比较,表 5列出以枞酸为基准的总能量、吉布斯自由能和焓这3个参数的相对值。
|
|
|
|
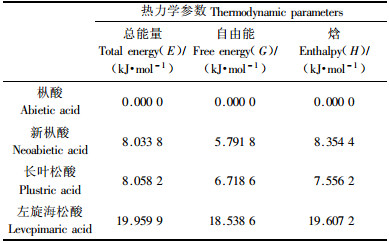
理论计算给出的总能量E和吉布斯自由能G的大小顺序为:枞酸>新枞酸>长叶松酸>左旋海松酸(表 4)。同时,表 5的理论热力学参数相对值显示,新枞酸和长叶松酸的总能量要比枞酸高出约8 kJ·mol-1,而左旋海松酸的则要高出枞酸约20 kJ·mol-1;吉布斯自由能的变化趋势也与总能量的趋势一样。根据最低能量原理,理论上得出4种主要树脂酸的相对稳定性次序为:枞酸>新枞酸>长叶松酸>左旋海松酸,理论计算结果与试验室测得的热稳定性顺序(枞酸>新枞酸>长叶松酸>左旋海松酸)(程芝,1996;沈美英等,2001)完全一致。说明精确的化学计算结果是合理和可信的。
3 结论本文对松香枞酸分子的可能构型进行了几何构型全优化,得到能量最低的最稳定的分子基态构型,并对最稳定的分子基态构型进行了几何参数比较分析。同时用密度泛函中的B3LYP泛函对枞酸、新枞酸、长叶松酸和左旋海松酸这4种树脂酸进行了能量分析。得到如下结果:
1) 就本文考察的体系而言,通过对几种化学计算方法得到的几何参数进行比较,发现从头算Hartree-Fock(HF)和密度泛函方法计算得到的几何参数都比半经验算法(MNDO)的更加接近试验的真实值,尤其密度泛函方法的计算结果与实际测量值的误差更小,因此B3LYP泛函更加适合用于对松香类化合物的分析与预测等方面的研究。
2) 本文用B3LYP泛函在6-31+G**机组水平下,得到枞酸、新枞酸、长叶松酸和左旋海松酸这4种树脂酸的热力学参数理论值,如总能量E、吉布斯自由能G、焓H、恒容热容C和熵S。对这几种树脂酸,理论计算结果预测的稳定性顺序与实验室测定的热稳定性顺序一致。
3) 本文运用B3LYP泛函的计算得到了比较满意的结果,考察的几何参数和热力学参数顺序与实验室的结果都有较好的正相关性,所以说本研究中B3LYP泛函的理论计算值是合理和可以接受的,结果对研究松香类化合物的晶体结构、热化学性质和反应性能有指导意义。
本文研究结果表明,计算化学方法也是科学研究的一种有用工具,精确的理论计算可以获得实验室难于得到的重要的化学与物理信息,用计算化学方法对松香类化合物进行模拟研究是可行的,这为该领域的研究引入了新的方法和内容,也为实验室研究松香类化合物提供理论依据,具有指导意义。
程芝. 1996. 天然树脂生产工艺学. 2版. 北京: 中国林业出版社, 107.
|
段文贵, 陈小鹏, 安鑫南. 2002. 氢化松香中性部分的化学组成. 南京林业大学学报:自然科学版, 26(2): 37-40. |
段文贵, 谢小光. 2003. 松香中枞酸型树脂酸的MNDO研究. 化学研究与应用, 15(2): 197-199. DOI:10.3969/j.issn.1004-1656.2003.02.014 |
沈美英, 梁忠云, 何正洪, 等. 2001. 湿地松树脂酸热异构反应的研究. 林产化工通讯, 35(2): 7-10. DOI:10.3969/j.issn.1673-5854.2001.02.002 |
宋湛谦. 2002. 松香的精细化工利用(Ⅰ)——松香的组成与性质. 林产化学通讯, 36(4): 29-34. |
张运明, 陈耀坤. 1995. 亚热带农产品的化学加工. 重庆: 重庆大学出版社, 113-114.
|
Becke A D. 1993. Density-functional thermochemistry Ⅲ. The role of exact exchange. J Chem Phys, 98: 5648-5652. DOI:10.1063/1.464913 |
Frisch M J, Pople J A, Binkley J S. 1984. Supplementary functions for Gaussian basis sets. J Chem Phys, 80: 3265-3269. DOI:10.1063/1.447079 |
Hohenberg P, Kohn W. 1964. Inhomogeneous electron gas. Phys Rev, 136: B864-B871. DOI:10.1103/PhysRev.136.B864 |
Kang Sunwoo, Yan Shihai, Lee Jin Yong. 2006. Theoretical studies on molecular recognition and self-assembly. Computing Letters, 2(4): 165-175. DOI:10.1163/157404006779194169 |
Kohn W, Sham L J. 1965. Self-consistent equations including exchange and correlation effects. Phys Rev, 140: A1133-A1138. DOI:10.1103/PhysRev.140.A1133 |
Legon A C. 1995. Interaction of CO and NH3 with BH3 and BF3. Chem Phys Lett, 237: 291-298. DOI:10.1016/0009-2614(95)00309-R |
Lenoir D, Chiappe C. 2003. What is the nature of the first-formed intermediates in the electrophilic halogenation of alkenes, alkynes and allenes?. J Chem Eur, 9: 1036-1044. DOI:10.1002/chem.200390097 |
Okada K, Takekuma S. 1994. Crystal Structure and conformational analysis of 7, 13-abietadien-18-oic acid. Bull Chem Soc Jpn, 67(3): 807-815. DOI:10.1246/bcsj.67.807 |
Pople J A, Gill P M W, Johnson B G. 1992. Kohn-sham density-functional theory within a finite basis set. Chem Phys Lett, 199: 557-561. DOI:10.1016/0009-2614(92)85009-Y |
Russo N, Toscano M, Grand A. 2003. Gas-phase theoretical prediction of the metal affinity of copper(Ⅰ) ion for DNA and RNA bases. J MassSpectrom, 38(3): 265-270. |
Tan Hongwei, Qu Wenwen, Chen Guangju, et al. 2003. Theoretical investigation of the self-assembly of cyclo[(-β3-HGly)4-]. Chemical Physics Letters, 369: 556-562. DOI:10.1016/S0009-2614(02)02031-6 |
Warren R M L, Tatehata A, Lappin A G. 1993. Effects of hydrogen bonding in electron-transfer reactions of tris (ethylenediamine) cobalt (2+). Inorg Chem, 32: 1191-1996. DOI:10.1021/ic00059a027 |
Wong Ming Wah. 1996. Vibrational frequency prediction using density functional theory. Chemical Physics Letters, 256: 391-399. DOI:10.1016/0009-2614(96)00483-6 |