 2006, Vol. 42
2006, Vol. 42文章信息
- 周祥明, 张冰玉, 苏晓华, 王大海, 黄秦军, 张香华, 张志毅.
- Zhou Xiangming, Zhang Bingyu, Su Xiaohua, Wang Dahai, Huang Qinjun, Zhang Xianghua, Zhang Zhiyi.
- 美洲黑杨雄性花芽cDNA克隆测序及表达序列标签(ESTs)特性分析
- Sequencing of cDNA Clones and Analysis of the Expressed Sequenced Tags(ESTs) Properties of Male Floral Buds of Populus deltoides
- 林业科学, 2006, 42(11): 37-41.
- Scientia Silvae Sinicae, 2006, 42(11): 37-41.
-
文章历史
- 收稿日期:2005-09-27
-
作者相关文章



2. 中国林业科学研究院林业研究所 国家林业局林木培育重点实验室 北京 100091
2. Key Laboratory of Tree Breeding and Cultivation, State Forestry Administration Research Institute of Forestry, CAF Beijing 100091
开花是植物从营养生长过渡到生殖生长的生理现象, 涉及到一个极为复杂的基因网络调控, 因此成为分子生物学研究的热点和难点。目前, 有关开花调控的分子遗传学研究多以1年生植物———拟南芥(Arabidopsis thaliana)为研究对象(Brunner et al., 2004), 而多年生木本植物可能采用了不同的进化途径, 在开花调控方面不同于1年生植物(Strauss et al., 2004)。因此, 以林木模式植物杨树为材料, 研究其开花相关的基因调控, 对深入研究植物开花调控机理具有重要意义。
cDNA部分测序是一种相对快速获取基因组中的未知特征基因表达和编码序列的方法(Adams et al., 1991)。cDNA即表达的基因只占基因组的2 %(Schuler, 1997), 长度一般为500~800 bp, 易于克隆和测序, 其主要策略是从来源于mRNA的cDNA文库中随机挑取克隆进行单向测序, 产生大量的部分cDNA约300~500 bp的短序列, 即ESTs(expressed sequence tags)序列(Adams et al., 1991; 陈亮等, 2005)。因此, ESTs技术现已被广泛应用于新基因的克隆、组织特异性基因表达谱分析、基因组序列的功能注释以及连锁图谱的构建等许多研究领域(Wu et al., 2002)。在杨树方面, 国外采用大规模的ESTs测序方法, 对白杨派树种形成层、叶柄、幼叶等不同组织器官的基因表达情况进行了研究(Fredrik et al., 2004), 获得了不同组织器官的特异表达基因及其表达谱, 但进一步的研究尚未见报道; 国内, 曾燕如等(2004)构建了杨树黑斑病cDNA文库。本研究以黑杨派的美洲黑杨(Populus deltoides)为研究材料, 构建国内第一个美洲黑杨雄性花芽cDNA文库, 并随机挑选4 200个经PCR检测过的单克隆进行大规模测序, 获得有效序列3 087个, 通过与核酸、蛋白数据库的比对、分析, 获得具有已知功能和未知功能的基因1 015个, 540个ESTs为新基因。这些数据为研究杨树及其他高等植物花发育提供了极有价值的资源。
1 材料与方法 1.1 cDNA文库构建以15年生雄性美洲黑杨雄花芽为材料, 采用SMART技术构建cDNA文库。构建方法参照SMART cDNA Library Construction Kit(Clontech #K1501-1)说明书进行。
1.2 cDNA克隆的大规模EST测序随机挑取经PCR检测含有插入片段的4 200个单克隆, 采用5'端测序, 测序仪为ABI PRISM 377。5'端测序引物为质粒pTriplEx2多克隆位点上游的一段序列, 其序列为5′-CTCCGAGATCTCGGACGAG-3′。
1.3 ESTs序列分析ESTs序列分析采用以下5个步骤: 1)对获得的序列采用Cross-match程序处理, 包括去除载体、去除短于150 bp的序列以及线粒体、叶绿体、rRNA等污染序列(污染标准: EST序列和污染序列库进行同源比对, 使用BLAST程序, evalue < 0.1, 当EST序列和目标序列的同源性大于50 %, 则认为该EST序列为污染序列)。2)采用BLAST工具包中的BLASTCLUST工具, 对经Cross-match程序处理过的数据进行聚类(聚类的标准:序列的相似区域长度超过序列的5 %并且该区域的相似度大于90 %)。3)对聚类后的每个Cluster进行单独拼接, 拼接采用CAP3, 得到相应的Contig和Singleton。4)将所有得到的Contig和Singleton合并, 通称Unique Sequence, 然后与SwissProt蛋白库(UniProt Release 5.3)作同源比对, 采用BLAST工具, evalue < 0.1, 同源比对相似度大于30 %的提取注释信息, 没有找到注释的序列进行功能域搜索, 采用InterPro工具包, 参数为默认参数, 仍然无法找到注释结果的, 认为是新基因(Novel Gene)。5)通过和SwissProt比对的acc number和interpro的结果, 对Unique Sequence进行GO分类(GO的版本为1.227, 更新时间为2005-06-21)。
2 结果与分析 2.1 cDNA文库质量检测随机挑选4 200个大肠杆菌(Escherichia coli)单菌落进行PCR扩增检测, PCR扩增产物片段长度范围为0.5~2.5 kb, 平均长度为1 kb。图 1为12个大肠杆菌PCR扩增结果, 其中第8泳道的PCR产物为200 bp, 根据试剂盒提供的载体序列说明, 判断是一个空载体, 未插入目的片段; 第3泳道出现2条带, 可能是模板受到污染; 其余PCR产物均为单一条带且大于500 bp, 说明已插入大小不同的目的片段, 同时也说明所构建的美洲黑杨雄性花芽cDNA文库是一个高质量的文库。

|
图 1 随机挑选12个大肠杆菌进行PCR检测 Fig. 1 PCR analysis of 12 randomly selected independent recombination of Escherichia coli M: Marker. |
从美洲黑杨雄性花芽cDNA文库中, 随机挑取经PCR检测过含有插入片段的4 200个单菌落进行测序, 获得原始序列3 092个, 测序成功率为73.6 %, 其中污染序列(线粒体、叶绿体、rRNA等)有6个(0.2 %), 有效序列为3 087个, ESTs的平均长度为515 bp。
对3 087个ESTs进行聚类分析, 获得416个聚类群(Clusters), 每个Cluster至少由2个认为是来源于同一个基因的ESTs组成。这些Clusters中含有1 983个ESTs, 其余1 104个为Singletons。冗余度(ESTs in clusters/total ESTs)为64 %, 表明如果在这个文库中继续进行随机选点测序, 还能发现新序列。
2.3 ESTs序列比对分析通过单一的EST序列(Unique Sequence of EST)与NCBI(National Center for Biotechnology Information)的非冗余核酸数据库的BlastN比对, 比对后的数据再与SwissProt蛋白库(UniProt Release 5.3)作同源比对, 采用BLAST工具, evalue < 0.1, 同源比对相似度大于30 %的提取注释信息, 并进行GO分类(Gene Ontology), 结果见表 1。
|
|
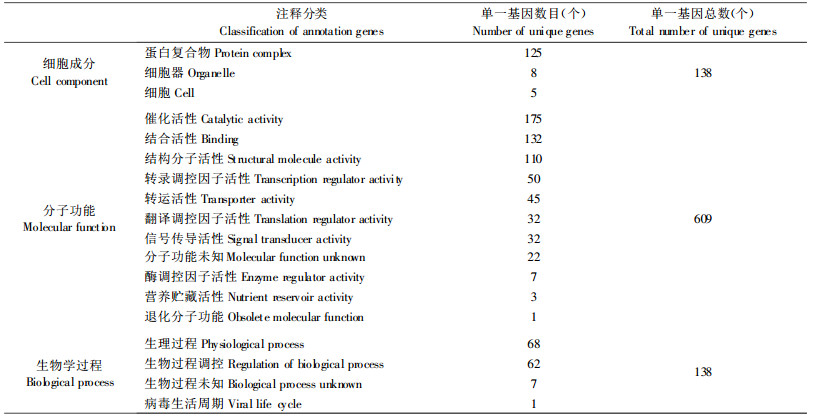
根据表 1中GO分类中的数据, 单一基因序列总数为885个。其中, 138个单一基因序列属于细胞成分基因序列, 占总单一基因的15.6 %; 具有分子功能的单一基因为609个, 占总单一基因的69 %; 比对结果属于生物过程的单一基因有138个, 占总单一基因的15.6 %。
与细胞成分相关的单一基因中, 蛋白复合物单一基因序列却占细胞成分基因的90.6 %, 占总单一基因的14 %。可能与取材时期材料生长发育活动旺盛有关。
在分子功能的单一基因中, 具有酶促活性、结合功能和结构分子活性的单一基因分别为175、132和110个, 而分子功能未知的单一基因有22个, 退化分子功能的单一基因只有1个。值得注意的是, 酶促活性、结合功能和结构分子活性的单一基因都是高丰度表达基因。植物表达的基因因组织、器官和发育阶段的不同而不同, 不同cDNA文库的基因具有不同的时空和组织特异性, 表达的丰度各异。与花发育相关的基因在这个时期都具有不同的表达丰度, 这些高丰度的表达基因可能与花发育相关。但同时, 次生代谢活动也旺盛, 这部分表达的基因丰度也高。
与生物过程有关的单一基因为138个, 其中生理过程和调节生物过程的基因分别为68和62个, 占总单一基因的8 %和7 %。图 2总结ESTs的GO分类比例。

|
图 2 ESTs的GO分类比例 Fig. 2 GO classification of identified ESTs according to putative gene function in percentage 1.蛋白复合物Protein complex; 2.细胞器Organelle; 3.细胞Cell; 4.催化活性Catalytic activity; 5.退化分子功能Obsolete molecular function; 6.结合活性Binding; 7.营养贮藏活性Nutrient reservoir activity; 8.结构分子活性Structural molecule activity; 9.转录调控因子活性Transcription regulator activity; 10.转运活性Transporter activity; 11.翻译调控因子活性Translation regulator activity; 12.信号传导活性Signal transducer activity; 13.分子功能未知Molecular function unknown; 14.酶调控因子活性Enzyme regulator activity; 15.病毒生活周期Viral life cycle; 16.生理过程Physiological process; 17.生物过程调控Regulation of biological process; 18.生物过程未知Biological process unknown. |
本文所获得的美洲黑杨雄性花芽ESTs数据对研究杨树花发育的分子机理以及克隆与花发育相关的基因具有重大作用。这个数据库中包含有开花相关的基因调控网络因子, 下一步结合基因芯片技术, 通过整体研究其调控开花的遗传网络, 可能比研究花发育网络调控中的单个基因更具实际意义, 但研究开花的整个遗传调控网络更具有挑战性。开花有5种途径:光质和光周期诱导途径、春化作用途径、自主途径、赤霉素(GA)途径、开花抑制途径, 这些途径相互作用, 构成了一个复杂的调控网络。Sheppard等(1997; 2000)和安新民(2004)研究花发育相关的单个基因在开花中的作用, 而未从整体的角度出发研究与花发育相关的所有基因。因此, 对花发育的ESTs分析, 有助于研究花发育相关基因的表达情况, 而且这个数据库对将来研究杨树性别决定和雄性不育都具有实际意义。
大规模ESTs测序已被证明是一种研究基因表达和寻找新基因的非常有效方法。从统计学角度分析, 为获得组织中主要表达基因, 至少需要1 000多条ESTs(Audic et al., 1997; 陈亮等, 2005)。本研究对美洲黑杨雄花芽cDNA文库的4 200个随机挑选克隆进行5'端测序, 去除载体序列, 平均插入片段长度515 bp。对ESTs分析而言, 这是一个有效长度(Hillier et al., 1996; Faccioli et al., 2001)。功能基因组序列只占基因组全序列的2 % (Schuler, 1997); 而某一时期表达基因一般只占全部基因的15 %左右, 因此ESTs分析研究功能基因组具有明显的优越性。其反映的是基因组的编码部分, 可以直接获得基因表达的信息, 跳过其他不表达的98 %“垃圾”序列(Schuler, 1997)。ESTs测序分析有助于探明植物生长、发育和分化(Sterky et al., 1998), 次生代谢和生化合成途径等分子机理(Lange et al., 2000; Mekhedov et al., 2000)。
为了获得高质量的序列, 去除了rRNA、叶绿体、线粒体等污染序列, 以及去除载体后插入片段长度小于150 bp的序列。将高质量的ESTs序列(3 087个)进行聚类, 获得416个clusters, 并对每个clusters进行单独拼接, 最后得到451个Contigs和1104个Singletons, 将1 555个ESTs与NCBI数据库进行比对, 其中34.7 %与核酸和蛋白数据库无序列同源性, 即34.7 %为新基因。虽然杨树的基因组测序已经完成, 但在基因功能和注释等方面还需要一定的时间, 因此出现34.7 %的基因在序列比对中, 无法提取相似的序列或注释, 在其他物种中也没有序列同源性, 说明这些基因在别的物种中尚未研究过。当然这个数据只是暂时的, 会随着研究的不断进行, 数据库信息的不断增加而减小。推测这些基因可能与杨树的开花调控相关。本课题若在资金与时间的允许下, 对这些新基因进行功能性研究, 可能会增加对杨树花发育的了解。我们所了解的有关植物开花时间调控来自对1年生的模式草本植物———拟南芥的研究(Mouradov et al., 2002; Simpson et al., 2002), 虽然大部分拟南芥基因家族在杨树中都有“副本”(Fredrik et al., 2004), 但它们的基因拷贝数不同, 而且在控制开花的多个方面也不同于拟南芥。下一步计划是结合基因芯片技术, 从整体上研究开花相关的网络调控, 为研究多年生木本植物开花机理奠定基础。
安新民.2004.毛白杨开花关键基因的分离及其功能分析.北京林业大学博士后出站报告
|
陈亮, 赵丽萍, 高其康. 2005. 茶树新梢cDNA克隆测序和表达序列标签(ESTs)特性分析. 农业生物技术学报, 13(1): 21-25. DOI:10.3969/j.issn.1674-7968.2005.01.004 |
曾燕如, 黄敏仁, 王明庥. 2004. 杨树黑斑病感抗无性系cDNA文库的构建. 南京林业大学学报:自然科学版, 28(3): 83-85. |
Adams M D, Kelley J M, Gocayne J D, et al. 1991. Comlpementary DNA sequencing : expressed sequence tags and human genome project. Science, 252: 1651-1656. DOI:10.1126/science.2047873 |
Audic S, Claverie J M. 1997. The significance of digital gene expression profiles. Genome Research, 7: 986-995. DOI:10.1101/gr.7.10.986 |
Brunner A M, Nilsson O. 2004. Revisiting tree maturation and floral initiation in the poplar functional genomics era. New Phytologist, 164: 43-51. DOI:10.1111/j.1469-8137.2004.01165.x |
Faccioli P, Pecchioni N, Cattivelli L, et al. 2001. Expressioned sequence tags from cold-acclimatized barley can identify novel plant genes. Plant Breeding, 120: 497-502. DOI:10.1046/j.1439-0523.2001.00652.x |
Fredrik S, Rupali R B, Unneberg P, et al. 2004. A Populus EST resource for plant functional genomics. Proc Natl Acad Sci USA, 101(38): 13951-13956. DOI:10.1073/pnas.0401641101 |
Hillier L D, Lennon G, Becker M, et al. 1996. Generation and analysis of 280, 000 human expressed sequence tags. Genome Res, 6: 807-828. DOI:10.1101/gr.6.9.807 |
Lange B M, Wildung M R, Stauber E J, et al. 2000. Probing essential oil biosynthesis and secretion by functional evaluation of expressed sequence tags from mint glandular trichomes. Proc Natl Acad Sci USA, 97: 2934-2939. DOI:10.1073/pnas.97.6.2934 |
Mekhedov S, Martinez O, Ohlrogge J. 2000. Towards a functional catalog of the plant genome : a survey of genes for lipid biosynthesis. Plant Physiology, 122: 389-401. DOI:10.1104/pp.122.2.389 |
Mouradov A, Cremer F, Coupland G. 2002. Control of flowering time interacting pathways as a basis for diversity. Plant Cell, 14: S111-S130. DOI:10.1105/tpc.001362 |
Schuler G D. 1997. Pieces of the puzzle:expressed sequence tags and the catalog of human genes. Journal of Molecular Medicine, 75: 694-698. DOI:10.1007/s001090050155 |
Sheppard L.1997. PTD : a Populus trichocarpa gene with homology to floral homeotic transcription factors. PhD thesis.Oregon State University, Corvallis, OR
|
Sheppard L A, Brunner A M, Krutovskii K V, et al. 2000. A DEFICIENS homolog from the dioecious tree black cottonwood is expressed in both female and male floral meristems of the wo-whorled, unisexual flowers. Plant Physiol, 124: 627-639. DOI:10.1104/pp.124.2.627 |
Simpson G G, Dean C. 2002. Arabidopsis, the Rosetta stone of flowering time?. Science, 296: 285-289. DOI:10.1126/science.296.5566.285 |
Sterky F, Regan S, Karlsson J, et al. 1998. Gene discovery in the wood-forming tissues of poplar : analysis of 5 692 expressed sequence tags. Proc Natl Acad Sci USA, 95: 13300-13335. |
Strauss S H, Martin F M. 2004. Poplar genomics comes of age. New Phytologist, 164: 1-4. DOI:10.1111/j.1469-8137.2004.01179.x |
Wu J, Maehara T, Yamamoto S, et al. 2002. A comprehensive rice transcript map containing 6 591 expressed sequence tag sites. The Plant Cell, 14: 525-535. DOI:10.1105/tpc.010274 |