 2006, Vol. 42
2006, Vol. 42文章信息
- 邹维华, 赵强, 崔德才, 王斌.
- Zou Weihua, Zhao Qiang, Cui Decai, Wang Bin.
- 反义磷脂酶Dγ基因与几丁质酶基因转化美洲黑杨G2
- Transformation of Populus deltoides with Anti-PLDγ Gene and Chitinase Gene
- 林业科学, 2006, 42(1): 37-42.
- Scientia Silvae Sinicae, 2006, 42(1): 37-42.
-
文章历史
- 收稿日期:2004-01-15
-
作者相关文章



2. 中国农业科学院生物技术研究所 北京 100081;
3. 中国科学院遗传与发育生物学研究所 北京 100101
2. Biotechnology Research Institute, Chinese Academy of Agricultural Sciences Beijing 100081;
3. Institute of Genetics and Developmental Biology, CAS Beijing 100101
利用基因工程技术选育耐盐杨树是开发利用盐碱地的一条十分有发展前景的途径,在防治土壤盐渍化和维护生态环境方面都具有十分重要的意义(刘岩等,1997)。在转入耐盐基因的基础上如能再将抗病基因转入杨树中,则会大大提高其开发利用的价值。本研究的目的就是试图将与耐盐有关的磷脂酶基因和与抗病有关的几丁质酶基因同时转入杨树中,以获得既耐盐又抗病的速生杨树新种质材料。
磷脂酶是通过调节细胞膜透性,影响细胞渗透压平衡和细胞信号转导的一类酶,与植物耐受逆境的能力直接相关,其中的磷脂酶D(PLD)在植物中广泛存在(Munnik et al., 1998;Wang, 1999)。研究表明,PLD不仅可以水解磷脂,改变细胞膜的稳定性和透性,而且水解产物中的磷脂酸、胆碱和乙醇胺等还是信号分子,参与细胞对外界环境刺激的应答过程和细胞内的信号传递,从而影响和调节细胞内一系列基因的表达(Wang, 1999;崔德才等, 2000)。在自然状态下,当植物受到逆境如病虫害、干旱、水淹等条件胁迫时,磷脂酶活性会在短时间内剧烈变化使膜的透性改变,从而引起植物的应答反应(Wang, 1997;1999;Lee et al., 1998;Munnik et al., 1998)。如果能在一定范围内抑制PLD的活性,则有可能改变植物对逆境的反应,减少伤害。对转PLDγ反义基因所得的拟南芥(Arabidopsis thaliana)和烟草(Nicotiana tabacum)的转化植株所做的测定表明,其PLD的活性明显受到抑制,抗逆性有了明显改变(Fan et al., 1997;1999)。刘斌等(2002)通过农杆菌介导法获得的转反义PLDγ基因的三倍体毛白杨(Populus tomentosa)BT-18,在实验室条件下显示其耐盐性明显高于对照,可在含68 mmol·L-1以上NaCl的培养基上正常生长。
几丁质酶具有降解几丁质(聚N-乙酰胺基葡萄糖)的作用(Linthort,1991)。许多危害植物的病原真菌的细胞壁主要成分之一是几丁质,而植物中还未发现几丁质酶的底物。所以,几丁质酶在防御病原真菌侵害中具有重要作用,它既能阻碍病原真菌细胞壁构成物质的沉积, 破坏细胞壁的正常建成, 又能降解病原真菌细胞壁中的几丁质,破坏真菌细胞壁, 而且这个过程产生的细胞壁碎片具有诱导物作用,从而刺激寄主植物产生抗病反应, 最终致使病原体死亡(Roberts et al., 1988)。转几丁质酶基因获得的烟草、水稻(Oryza sativa)、小麦(Triticum aestivum)等抗病性都有所提高(Broglie et al., 1991; Villie,1995; Li et al., 2001)。
本研究选用单基因连续转化法,通过农杆菌介导先把反义磷脂酶Dγ基因转到美洲黑杨(Populus deltoides)G2中,获得耐盐植株后,再转入几丁质酶基因,最终获得了转双价基因的G2速生黑杨新种质材料。
1 材料与方法 1.1 试验材料与菌种美洲黑杨G2为一优良无性系,由中国科学院遗传研究所与格林公司、北京农林科学院联合培育,中国科学院遗传与发育生物学研究所泰安生物技术实验基地提供。PLDγ反义基因由美国肯萨斯州立大学王学敏教授惠赠,载体质粒为pKYLX7(见图 1),农杆菌菌株为EHA105,Anti-PLDγ基因由花椰菜花叶病毒的35S启动子驱动,含新霉素磷酸转移酶基因,具卡那霉素抗性,终止子为二磷酸核酮糖羧化酶小亚基E9基因3'末端终止子rbcS3'。菜豆几丁质酶基因CH5B载体质粒为p1300CH5B,由王斌实验室克隆并保存,CH5B基因由双35S启动子驱动,几丁质酶基因后连有GUS报告基因,具潮霉素抗性(见图 2)。

|
图 1 质粒pKYLX7的结构 Fig. 1 Diagram of plant expression vector pKYLX7 |

|
图 2 质粒p1300CH5B的结构 Fig. 2 Diagram of plant expression vector p1300CH5B |
为确定G2杨对卡那霉素及潮霉素的抗性,将生长健壮的幼嫩叶片及高约3~5 cm的芽分别接种于含不同浓度抗生素的叶分化培养基和生根培养基上,进行叶片诱导分化及芽诱导生根的抗生素临界浓度试验。
1.2.2 农杆菌介导的杨树叶盘转化改良的农杆菌介导杨树叶盘转化法参照刘斌等(2002)的方法进行,通过延长共培养时间提高了叶片转化率,转化叶片在筛选培养基上生长3~4周后,转移到含筛选抗生素的生根培养基上,将能正常生根的株系扩繁作为分子检测的材料。
1.2.3 杨树再生苗的PCR检测采用SDS微量提取法(王关林等,2002)从幼嫩组培苗的叶片中提取植株总DNA, 取100~500 ng, 分别用反义PLDγ及几丁质酶基因的引物, 参照Carl等(1995)的方法进行PCR检测,按标准反应程序进行PCR扩增,PCR产物经1.2%琼脂糖凝胶电泳、EB染色后, 在UV灯下观察结果。
1.2.4 转基因杨树的PCR-Southern杂交鉴定PCR-Southern杂交参照Sambrook等(1989)的方法进行,PCR产物经1.2%琼脂糖凝胶电泳后,用毛细管转移法转移到Hybond-N+尼龙膜上,预杂交,杂交后进行放射性自显影。探针用随机引物法合成,采用Promega的Primer-a-Gene Labeling System试剂盒,试验中用转化质粒DNA作为阳性对照,用未转基因的美洲黑杨G2植株DNA作为阴性对照。
1.2.5 转反义PLDγ基因植株的耐盐性鉴定取株高3~5 cm,转反义PLDγ基因PCR-Southern杂交阳性植株和未转化的阴性对照植株各10株,接种于不同含盐量生根培养基上,进行耐盐性分析。NaCl浓度分别为0,68, 102, 136, 170 mmol·L-1,18 d后检测结果。
1.2.6 GUS基因稳定表达的组织化学染色GUS基因稳定表达的组织化学染色分析参照王关林等(2002)的方法进行,取转几丁质酶基因PCR-Southern杂交阳性植株和未转化的阴性对照植株叶片3~4片浸泡在GUS染色液中于37 ℃保温过夜,将材料转入70%的乙醇中脱色,至阴性对照材料呈白色,肉眼观察拍照。
2 结果 2.1 卡那霉素(Km)和潮霉素(Hyg)临界筛选浓度结果表明,低浓度的卡那霉素(Km)即抑制叶片产生不定芽。在不含Km的对照培养基上,外植体产生大量丛生芽,当Km浓度大于20 mg·L-1时,叶片既不分化出芽也不产生愈伤组织。故Km 20 mg·L-1即可作为临界浓度筛选转化细胞。叶片分化对潮霉素非常敏感,5 mg·L-1时叶片即不能分化,10 mg·L-1时叶片变褐,坏死,本试验选取10 mg·L-1筛选转化细胞。
芽诱导生根的临界卡那霉素浓度要高于叶分化所需浓度,Km浓度大于30 mg·L-1时就没有根分化发生,因此取30 mg·L-1的卡那霉素筛选抗性植株。芽诱导生根对潮霉素也非常敏感,5 mg·L-1时即不能生根,与培养基接触部位变白,叶片变黄。试验中取10 mg·L-1筛选转化植株。
2.2 转基因植株的抗性筛选转反义PLDγ基因的叶片在附加头孢霉素(Cef)400 mg·L-1、卡那霉素(Km)20 mg·L-1的分化培养基上20 d后即分化出抗性芽,待小芽长至0.5 cm左右时转到含相同抗生素浓度的扩繁培养基上,15 d转接1次,转接3次,其中大多数小芽黄化死亡。将获得的抗性芽转到附加Cef 400 mg·L-1,Km 30 mg·L-1的生根培养基上,十几天后即可生根。获得了21株卡那霉素抗性植株,转化植株扩繁后进行分子检测。
转CH5B基因的叶片经共培养10 d,待叶片上有芽点突起后再将叶片转移到附加Cef 400 mg·L-1、潮霉素(Hyg)10 mg·L-1的分化培养基上,培养期内叶片严重失绿褐化,仅局部可见绿色芽点分化长大。25 d后转至含相同抗生素的扩繁培养基上继续培养30 d以上,芽丛生长缓慢,仅长至0.5 cm大小。将芽丛转至不含潮霉素仅附加头孢霉素的培养基上扩繁,待芽长至3~5 cm时进行抗生素生根培养,在含Hyg 10 mg·L-1的培养基上10 d后有6株正常生根,将这6个株系分别扩繁,进行分子检测。
考虑到美洲黑杨本身含有PLDγ基因,所以在引物设计上做了些调整。反义PLDγ基因5'引物从35S启动子中选取,3'引物从PLDγ反义基因内部选取,以含反义PLDγ基因的质粒pKYLX7作阳性对照,以未转基因的美洲黑杨G2植株作为阴性对照,进行PCR扩增。13株卡那霉素抗性植株呈阳性,5株卡那霉素抗性植株PCR结果见图 3,其余8株PCR图略。转基因的植株得到一条和预期长度一样大小的400 bp的目的片段,有的样品除目的带外,还扩增出了非特异带,未转基因植株也有此非特异带扩增,优化PCR条件后依然如此,这可能是内源PLD非特异性扩增的结果。为了进一步证明PCR扩增结果,将PCR扩增产物电泳后转印于尼龙膜上,进行PCR-Southern杂交检测(图 4)。PCR阳性植株扩增产物目的带显示出较强信号,非特异带没有显示杂交信号,对照植株没有显示信号,说明反义PLDγ基因已整合到美洲黑杨G2基因组中。

|
图 3 反义PLDγ基因的PCR检测 Fig. 3 PCR assay of anti-PLDγ gene 1:2 kb标准物2 kb lader; 5:质粒DNA(阳性对照) Plasmid DNA as positive control; 8:非转基因植株(阴性对照)Non-transformed plant as negative control; 2, 3, 4, 6, 7:转化植株Transgenic plants.箭头指出400 bp的目的扩增片段Arrow indicates the target amplification fragment. |

|
图 4 转反义PLDγ基因PCR-Southern杂交检测 Fig. 4 PCR-Southern hybridization assay of anti-PLDγ gene 1, 2, 3, 5, 6:转基因植株Transgenic plants; 4:质粒DNA(阳性对照) Plasmid DNA as positive control; 7:非转基因植株(阴性对照)Non-transformed plant as negative control.2.3 PCR与PCR-Southern鉴定 |
转几丁质酶基因的潮霉素抗性植株的PCR检测结果如图 5所示。6个抗性株系全表现为阳性,经PCR-Southern杂交检测显示较强信号(图 6),而阴性对照既无PCR扩增带也无PCR-Southern杂交信号。证明CH5B基因已整合到美洲黑杨G2基因组内。

|
图 5 转几丁质酶基因的PCR检测 Fig. 5 PCR assay of CH5B gene 1:2 kb标准物2 kb lader; 2, 4, 5, 6:p1300CH5B质粒DNA(阳性对照) p1300CH5B plasmid DNA as positive control; 3:非转基因植株(阴性对照) Non-transformed plant as negative control; 7~12:转化植株Transgenic plants.箭头指出目的扩增片段Arrow indicates the target amplification fragment. |

|
图 6 转几丁质酶基因PCR-Southern检测 Fig. 6 PCR-Southern assay of CH5B gene 1:阳性对照Positive control; 2~7:转基因植株Transgenic plants.箭头指出目的扩增片段Arrow indicates the target amplification fragment. |
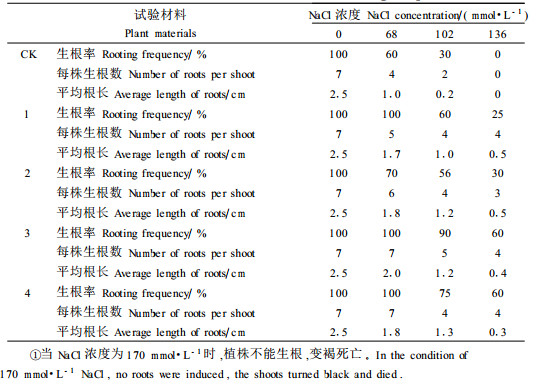
取株高3~5 cm的转基因无根试管苗,接种于含不同NaCl浓度的生根培养基上,进行耐盐性分析。培养18 d后进行统计分析,结果见表 1。对照植株在含NaCl 136 mmol·L-1以上的培养基上不能生根,叶子由灰黄变白变黑,植株全部死亡。13株PCR-Southern阳性植株则表现不一,从中筛选到4株耐盐性强的植株。这4个植株及其扩繁的后代在NaCl浓度为0~68 mmol·L-1的培养基上生长良好,叶子鲜绿色。随NaCl浓度升高,植株生长受抑程度逐渐加深,在含136 mmol· L-1 NaCl的生根培养基上仍能够生根生长,根长0.3~0.5 cm。在170 mmol·L-1 NaCl的生根培养基上不能生根,植株变褐死亡。由此可以证明,转PLDγ反义基因确实明显提高了美洲黑杨G2的耐盐性。
|
|
由于CH5B基因后连有报告基因GUS,处于同一开放阅读框内,因此通过检测GUS基因的稳定表达,就可以证明几丁质酶基因获得表达。GUS基因稳定表达的组织化学染色表明,所获6个转基因植株及其后代中的GUS基因正常表达,同时可以推断CH5B基因也能表达。
3 讨论随着植物基因工程技术的日益成熟和完善,为了使受体植物获得多个优良新性状,现代基因转化的趋势往往是将2个或2个以上的基因转入同一个受体之中,如在抗病基因工程中常常转入多种生化底物的抗菌蛋白,以使植物获得抵抗多种病原物侵害的能力。同样在生产某一代谢物时,也需要转入该代谢物生化合成途径中的多个基因。多基因转化植株的获得有以下几种途径:
第1种是分步进行单个基因的转化,再进行转基因植株的杂交(Zhu et al., 1994; Bizily et al., 2000)。此方法的缺陷是后代中不同的转基因位于不同的基因位点,使杂交后的育种过程变得非常复杂,而且这种方法不适用于马铃薯(Solanum tuberosum)、甘薯(Ipomoea batatas)、果树、林木等无性繁殖植物。
第2种是单基因连续转化法,即先转入一个基因,获得转基因植株后再转入另一个基因。此方法需要在各步的基因转化过程中使用不同的选择标记。
第3种是将多个基因构建于一个转化载体中,基因间首尾相连接,各个基因都有自己的启动子与终止子(Slater et al., 1999)。这是目前广泛采用的一种方法。有试验表明,同一转基因植株内多拷贝的同一启动子的存在易导致转基因沉默(Van et al., 1993),因此要选用不同的启动子,而找到一套在细胞类型、表达水平、发育特异性及对环境应答等方面表现一致的启动子又变得非常困难。
第4种是基于由一个转录单元编码的多蛋白前体经蛋白酶剪切后能产生多种蛋白质的机制(Carrington et al., 1988),将多个基因构建于同一表达框下,基因间通过连接子(linker)相连,该连接子可被体内蛋白酶切割。这种方法是当前研究的热点,但由于缺乏对蛋白质剪切机制的深入了解,构建的多个载体转化后不能被完全切割,产生的蛋白质往往携带连接子的几个氨基酸残基,使蛋白质功能大大下降(Urwin et al., 1998;Halpin et al., 1999;Isabelle et al., 2002)。
综合以上几种方案,本研究采用第2种方案,即首先通过卡那霉素筛选获得转反义PLDγ基因植株, 经耐盐性筛选获得高耐盐植株后,继续转入几丁质酶基因,通过潮霉素筛选获得转化植株,由于每个基因转化之后都进行了严格的抗生素筛选和分子鉴定,因此那些非转化植株及表达较弱的植株都已被淘汰,筛选得到的是反义PLDγ和几丁质酶基因整合及表达都是最好的植株。
众多资料表明,植物的耐盐性是多种抗盐生理性状的综合表现,是由位于不同染色体上的多个基因控制的(邱德有等,1993)。利用转基因技术获得的转基因植株虽有一定的抗盐性,但这只是相对于对照株而言的,因此,利用转基因技术研究植物的抗盐性还需要深入的研究。反义PLDγ基因的导入使部分转化植株的耐盐性有所提高,还只是实验室内获得的初步结果,今后将结合田间试验进一步加强植物耐盐机制的研究。至于转基因植株的抗病性还有待测定,如进行几丁质酶活性测定和病原菌接菌试验。杨树为多年生木本植物,其耐盐性及抗病性的鉴定要经过3~5年,甚至更长时间的田间实地栽培才能获得比较肯定的结果。
崔德才, 温孚江. 2000. 磷脂酶D(PLD)在植物信号转导中的作用. 山东农业大学学报:自然科学版, 31(2): 115-119. |
刘斌, 李红双, 王其会, 等. 2002. 反义磷脂酶Dγ基因转化毛白杨的研究. 遗传, 24(1): 40-44. |
刘岩, 彭学贤, 谢友菊, 等. 1997. 植物抗渗透胁迫基因工程研究进展. 生物工程进展, 17(2): 31-38. |
邱德有, 朱徵. 1993. 植物抗渗透胁迫的基因调控及基因工程. 生命科学, 5(4): 16-17. |
王关林, 方宏筠. 2002. 植物基因工程. 2版. 北京: 科学出版社.
|
Bizily S P, Rugh C L, Meagher R B. 2000. Phytodetoxification of hazardous organomercurials by genetically engineered plants. Nat Biotechnol, 18: 213-217. DOI:10.1038/72678 |
Broglie K, Chet I, Holliday M. 1991. Transgenic plants with enhanced resistance to the fungal pathogen Rhizoctonia solani. Science, 254: 1194-1197. DOI:10.1126/science.254.5035.1194 |
Carl W D, Gabriela S D. 1995. PCR primer: A laboratory manual. New York: Cold Spring Harbor Laboratory Press.
|
Carrington J C, Dougherty W G. 1988. A viral cleavage site cassette: identification of amino acid sequences required for tobacco etch virus polyprotein processing. Proc Natl Acad Sci USA, 85: 3391-3395. DOI:10.1073/pnas.85.10.3391 |
Fan L, Zheng S Q, Wang X M. 1997. Antisense suppression of phospholipase D retard abscisic acid and ethylene-promoted senescence of postharvest Arabidopsis leaves. The Plant Cell, 9: 2183-2196. |
Fan L, Zheng S Q, Cui D C, et al. 1999. Subcellular distribution and tissue expression of phospholipase Dα, Dβ and Dγ in Arabidopsis. Plant Physiol, 119: 1371-1378. DOI:10.1104/pp.119.4.1371 |
Halpin C, Cooke S E, Barakate A, et al. 1999. Self-processing 2A-polyproteins: a system for coordinate expression of multiple proteins in transgenic plants. Plant J, 17: 453-459. DOI:10.1046/j.1365-313X.1999.00394.x |
Isabelle E J A F, Miguel F C D, Geoff Dwyer, et al. 2002. Transgenic expression in Arabidopsis of a polyprotein construct leading to production of two different antimicrobial proteins. Plant Physiology, 128: 1346-1358. DOI:10.1104/pp.010794 |
Lee S H, Chae H S, Lee T K, et al. 1998. Ethylene mediated phospholipid catabolism pathway in glucose-starved carrot suspension cells. Plant Physiology, 116: 223-229. DOI:10.1104/pp.116.1.223 |
Li W L, Faris J D, Muthukrishnan S, et al. 2001. Isolation and characterization of novel cDNA clones of acidic chitinases and β-1, 3-glucanases from wheat spikes infected by Fusarium graminearum. Theor Appl Genet, 102: 353-362. DOI:10.1007/s001220051653 |
Linthort H J M. 1991. Pathogenesis-related proteins of plants. Critical reviews in Plant Sciences, 10: 123-150. DOI:10.1080/07352689109382309 |
Munnik T, Irvine R F, Musgrave A. 1998. Phospholipid signaling in plants. Biochemical et Biophysical Acta, 1389: 222-272. DOI:10.1016/S0005-2760(97)00158-6 |
Roberts W K, Selitrennikoff C P. 1988. Plant and bacterial chitinases differ in antifungal activity. J of Gen Microbiol, 134: 169-765. |
Sambrook J, Fritsch E F, Maniatis T. 1989. Molecular cloning: A laboratory manual. 2nd ed. New York: Cold Spring Harbor Laboratory Press.
|
Slater S, Mitsky T A, Houmiel K L, et al. 1999. Metabolic engineering of Arabidopsis and Brassica for poly 3-hydroxybutyate-co-3-hydroxyvalerate copolymer production. Nat Biotechnol, 17: 1011-1016. DOI:10.1038/13711 |
Urwin P E, McPherson M J, Atkinson H J. 1998. Enhanced transgenic plant resistance to nematodes by dual proteinase inhibitor constructs. Planta, 204: 472-479. DOI:10.1007/s004250050281 |
Van den Elzen P J M, Jongedijk E, Melchers L S, et al. 1993. Virus and fungal resistance: from laboratory to field. Phil Trans R Soc Lond B, 342: 271-278. DOI:10.1098/rstb.1993.0157 |
Villie L. 1995. Genetic engineering of rice for resistance to sheath blight. Biotechnology, 13: 686-691. |
Wang X M. 1997. Molecular analysis of phospholipase D. Trends in Plant Science, 2(7): 261-266. DOI:10.1016/S1360-1385(97)86348-0 |
Wang X M. 1999. The role of phospholipase D in signal transduction cascade. Plant Physiology, 120: 456-462. |
Zhu Q, Maher E A, Masoud S, et al. 1994. Enhanced protection against fungal attack by constitutive co-expression of chitinase and glucanase genes in transgenic tobacco. Biotechnology, 12: 807-812. |