 2009, Vol. 45
2009, Vol. 45文章信息
- 徐煲铧, 杨晓慧, 李百炼, 张志毅, 张德强.
- Xu Baohua, Yang Xiaohui, Li Bailian, Zhang Zhiyi, Zhang Deqiang
- 毛白杨纤维素合酶基因PtCesA4的克隆、表达及单核苷酸多态性分析
- Isolation, Expression and Single Nucleotide Polymorphisms Analysis of Cellulose Synthase gene (PtCesA4) from Populus tomentosa
- 林业科学, 2009, 45(5): 1-10.
- Scientia Silvae Sinicae, 2009, 45(5): 1-10.
-
文章历史
- 收稿日期:2008-12-19
-
作者相关文章



2. 北京林业大学林木育种国家工程实验室 北京 100083;
3. 美国北卡罗莱纳州立大学林学系 北卡罗莱纳州 NC27695-8203
2. National Engineering Laboratory for Tree Breeding, Beijing Forestry University Beijing 100083;
3. Department of Forestry, North Carolina State University North Carolina State 27695-8203
纤维素是木材的主要组成物,是自然界最重要的可再生工业原料。在树木中,纤维素含量的多少将直接影响到发酵产物乙醇的转化率和制浆造纸过程中纸浆得率(Chiang et al., 1988;Sassner et al., 2005)。因此,深入研究纤维素的生物合成过程,了解控制木材纤维品质的基因是对其进行遗传操作的基础。为此,先前的研究者对纤维素的生物合成过程进行了广泛研究,认为在生物体内纤维素主要由纤维素合酶催化合成β-D-吡喃葡萄糖单元并通过(1—4)糖苷键连接而成为高聚糖最终形成纤维素(Kimura et al., 2002)。纤维素合酶基因(cellulose synthase,CesA)首先由Saxena等(1990),从木醋杆菌(Acetobacter xylinum)中克隆和鉴定,随后Saxena等(1995)将该基因与其他糖苷转移酶的氨基酸序列进行了比较分析,结果发现其具有β-糖苷转移酶的一些特征,如包括8个转膜区域和非常保守的DDDQxxRW(天冬氨酸,天冬氨酸,天冬氨酸,谷氨酰胺-x-x-精氨酸-色氨酸)序列,认为纤维素合酶是一种整合性的质膜蛋白。借助细菌纤维素合酶的基因序列,Pear等(1996)在分析棉花(Gossypium hirsutum)纤维表达序列标签(expressed sequence tags,ESTs)时首次发现2个植物纤维素合酶基因,并将其命名为GhCesA-1与GhCesA-2;表达分析显示:在棉花纤维形成过程中,这2个基因在次生细胞壁纤维素合成时高水平表达,据此可推测它们可能编码棉花纤维素合酶蛋白。其后,对基因组和EST序列分析表明,在1年生植物如拟南芥(Arabidopsis thaliana)、水稻(Oryza sativa)和大麦(Hordeum vulgare)等植物中至少存在10个CesA基因(Richmond et al., 2000;Tanaka et al., 2003;Burton et al., 2004)。
鉴于树木纤维素对林产工业经济价值的重要性,近年来Wu等(2000)从模式树木美洲山杨(Populus tremuloides)中分离出第1个树木CesA基因,表达分析初步显示该基因在正在发育的木质部组织中特异表达。最近,毛果杨(P.trichocarpa)全基因组测序工作的完成和许多可利用的杨树EST数据库为检测和克隆纤维素合酶基因家族所有成员提供了可能。Djerbi等(2005)利用生物信息学手段,以拟南芥CesA基因的核苷酸序列和氨基酸序列为信息探针,对杨树全基因组进行扫描并结合EST数据库,检测到了18个杨树CesA基因成员,认为在杨树中至少存在18个CesA基因成员。虽然对纤维素的生物合成过程及其调控机制有了初步认识,但不同树种纤维素生物合成的基因调控机制不尽相同,仍需要进行纤维素合酶基因的克隆和检测。特别是最近产生的基于候选基因内单核苷酸(single nucleotide polymorphisms,简称SNPs)的联合遗传学研究对于林木育种方案的制定非常有用(Zhang et al., 2005)。而毛白杨(P.tomentosa)是我国特有的杨属白杨派树种,分布在我国黄淮海流域约100万km2的范围内,在我国北方,尤其在黄河流域林业生产和生态环境建设中占有重要地位。在长期进化过程中,在毛白杨种内形成了许多变异类型,而这种积累的变异能被用来研究控制目标性状的分子遗传学基础(朱之悌,2005)。鉴于毛白杨野生性强、遗传变异丰富、进化史较长等特点,可能很适合用于开展林木数量性状相关基因的SNP发现和联合遗传学研究。为此,本研究从毛白杨未成熟木质部cDNA中分离出一纤维素合酶基因,并对其进行了组织特异性表达、单核苷酸多态性和连锁不平衡分析。研究结果为分离其他树种CesA成员以及进行该基因的连锁不平衡(linkage disequilibrium,简称LD)作图和基因辅助树木育种提供了重要的依据。
1 材料与方法 1.1 植物材料1983—1984年,全国毛白杨协作组从毛白杨分布区如北京、河北、山东、河南、山西、陕西、甘肃、宁夏、安徽和江苏10个省(市、自治区)100个种源,选取了1 047株基因型个体,通过根繁方式在山东省冠县国营苗圃建立了全国毛白杨基因库。依据这100个种源所反映的热、光和水等16个气象因子,通过主分量分析,将整个毛白杨分布区划分为3个气候区,即东北部气候区(包括北京、河北和山东)、南部气候区(河南、安徽和江苏)和西北气候区(山西、陕西和甘肃),经F检验这3个气候区的差异达显著水平(朱之悌,2005)。本研究提取DNA的叶片材料取自该基因库中能够最大程度地反映毛白杨分布范围的40个种源,即40株基因型个体,并置于液氮中冻存备用。
提取RNA的材料取自中国林业科学研究院院内毛白杨雌株的形成层组织并置于液氮中冻存备用。
1.2 总DNA的提取总DNA的提取按DNeasy Plant Mini Kits (Qiagen,Inc.,Valencia,CA,USA)描述的方法进行。
1.3 RNA提取和cDNA的合成毛白杨根部组织、树皮、韧皮部、形成层、未成熟木质部、成熟木质部、成熟叶片和顶端分生组织材料总RNA的提取和cDNA的合成分别按RNeasy Plant Mini Kits (Qiagen,Inc.,Valencia,CA,USA)和Clonetech试剂盒描述的方法进行。
1.4 电子杂交以拟南芥AtCesA4的氨基酸序列(GenBank注册号为NP_199216)为信息探针,将其输入瑞典和法国杨树EST数据库(Sterky et al., 2004)以及毛果杨全基因组数据库(Wullschleger et al., 2002)同源比较服务器进行BLASTX分析(Altschul et al., 1997),筛选出蛋白质氨基酸序列同源性在75%以上的ESTs和Contigs作为候选序列,然后逐一将这些序列送GenBank核酸数据库检索,后将这些序列进行整合拼接,获得统计上完整的序列。
1.5 引物设计和PCR扩增根据所得的EST重叠群整合序列,在其可能的开放阅读框两端设计一对寡聚核苷酸引物:PtCesA4F:5′-GGTATCTGAAACACCTTTGAGTCTAAGCGA-3′;PtCesA4R:5′-AGAGGGATAGATTCCTTGCCTTGACCTGGA-3′。
应用25 μL DNA聚合酶链式反应(PCR)体系,以毛白杨未成熟木质部cDNA为模板,加入2.5 μL 10 × buffer,1.8 μL 25 mmol·L-1 MgCl2,1 μL 10 mmol·L-1 dNTP,Taq DNA聚合酶1.0 U(以上试剂购自Promega公司),100 nmol·L-1正向和反向引物各1 μL,加适量双蒸水至25 μL。于94 ℃,3 min →(93℃,30 s →65 ℃,30 s→72 ℃,1 min) 40个循环→72 ℃,5 min热循环条件下,扩增出长约3800 bp的cDNA片段。
1.6 PCR产物的克隆、测序及计算机分析将PCR扩增得到的目的基因片段回收后连接于pGEM-T上。连接产物转化大肠杆菌DH5α,筛选阳性克隆,送公司测序。应用DNAMAN 6.0软件和CLUSTALX软件包程序推导出氨基酸序列并进行NCBI检索分析,然后分别估算和预测推导蛋白质的分子质量和等电点。
1.7 Realtime PCR检测PtCesA4基因在不同组织和器官中的表达模式采用ABI Primer Express 3.0软件设计能够扩增长度约为100 bp cDNA片段的Realtime PCR引物,其序列如下:
PtCesA4RTF:5′-GCCAGTCTGCAACGTCGAA-3′;
PtCesA4RTR:5′-GGAAAGCCACACACATGAC-3′。
以肌动蛋白基因Actin作为内参,引物序列为ActF:5′-CTC,CAT,CAT,GAA,ATG,CGA,TG-3′;ActR:5′-TTG,GGG,CTA,GTG,CTG,AGA,TT-3′。定量PCR反应按照SYBR® RPREMIX EX TAQTM (TAKARA)试剂盒描述的方法进行。每个PCR 8连管中设置4个样品重复、4个内对照重复。依次加入SYBR® RPREMIX EX TAQTM 12.5 μL(2×),定量PCR上游引物0.5 μL (10 μmol·L-1),下游引物0.5 μL (10 μmol·L-1),cDNA 2 μL,终体积为25 μL,轻微离心,收集到管底。PCR反应在MJ Research Opticon 2荧光检测系统上进行Real-time PCR反应,其扩增反应程序为:94 ℃,5 min→(94 ℃,30 s→58 ℃,30 s→ 72 ℃,30 s) 40个循环。PCR结束后,制作融解曲线检查是否有非特异扩增,然后应用MJ Research Opticon 2 SYSTEM软件分析结果。
1.8 PtCesA4基因的SNP检测依据已克隆的PtCesA4基因的核苷酸序列设计基因特异的引物,以选取的40株个体的总DNA为模板进行PCR扩增;将PCR扩增产物进行琼脂糖凝胶电泳分离,回收、纯化目的片段后与PGEM-T载体连接,转化后挑取阳性单克隆进行序列测定,然后将每一基因片段的核苷酸序列拼接成完整的基因序列。
1.9 PtCesA4基因的SNP多样性分析和连锁不平衡检测利用MEGA4.50.4软件对PtCesA4基因的40个序列进行比对分析,标出SNP位点,计算SNP频率、转换和颠换的SNP数量;计算SNP多样性指数(π,任意两序列之间核苷酸差异的平均数除以核苷酸总数)、在基因不同区域的分布模式;分析同义突变、错义突变和无义突变以及发生在剪切位点(GT-AG)的突变情况;分析LD在不同基因中的延伸模式。
2 结果与讨论 2.1 毛白杨PtCesA4同源cDNA的克隆及其结构特征分析为了分离杨树CesA4基因的cDNA序列,从拟南芥AtCesA4氨基酸序列出发,筛选杨树ESTs和毛果杨全基因组数据库。对于CesA4基因,利用电子杂交的策略在毛果杨全基因组上获得了同源性高达75%以上的1个CesA4候选序列。利用设计的引物PtCesA4F和PtCesA4R,以毛白杨未成熟木质部总cDNA为模板进行PCR扩增。扩增产物经0.8%琼脂糖凝胶电泳分离,在大小约3 800 bp处扩增出1条明亮的特异条带(图 1)。将扩增的特异片段回收、纯化并与pGEM-T载体连接后克隆测序。测序结果表明,克隆的这一PtCesA4 cDNA总长为3 757 bp,在PtCesA4序列中,基因内部含有完整的开放阅读框架,大小为3 129 bp,可编码长度为1 042个氨基酸残基的蛋白质,并将其进行GenBank提交和注册,其序列接收号为FJ534554。在此基础上,运用DNAMAN6.0软件估算推导的PtCesA4蛋白质的分子质量约为118.4 ku,其等电点为7.6。

|
图 1 从毛白杨未成熟木质部cDNA中扩增的PtCesA4 cDNA片段 Figure 1 The cDNA fragment of PtCesA4 amplified from immature xylem cDNA in P.tomentosa M:1 kb DNA ladder;1:从毛白杨未成熟木质部cDNA中扩增的PtCesA4 cDNA片段 The cDNA fragment of PtCesA4 amplified from immature xylem cDNA in P.tomentosa. |
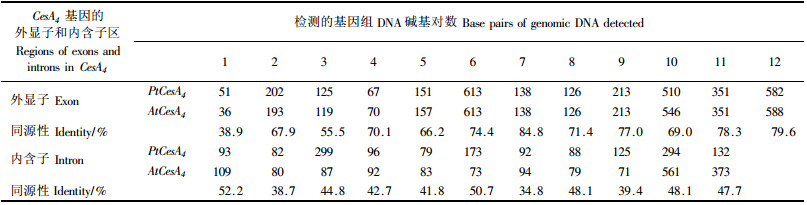
在克隆PtCesA4 cDNA的基础上,将其核苷酸序列与模式植物拟南芥基因组的AtCesA1—AtCesA10共10个纤维素酶cDNA序列进行了同源性比对分析,结果显示,克隆的毛白杨PtCesA4与拟南芥CesA基因高度同源,同源性由63.9%(AtCesA10)~73.6%(AtCesA4)。因此,将克隆的这一毛白杨CesA命名为PtCesA4是合理的。为了进行木本植物与草本植物CesA4基因组结构的比较基因组学研究,以毛白杨基因组总DNA为模板进行了PCR扩增和序列测定,并将其与对应的拟南芥AtCesA4基因的外显子和内含子长度及同源性进行了比较(表 1)。由表 1可见,毛白杨PtCesA4与拟南芥AtCesA4编码区所覆盖的基因组总长度分别为4 682 bp和4 852 bp,它们具有相同的基因组结构,即都含有12个外显子(Exon)和11个内含子(Intron)。虽然木本植物与草本植物CesA4基因的总长度相差170 bp,但其Exon长度却非常接近,特别是Exon 6—Exon 9和Exon 11的长度完全相同,即均含有613,138,126,213,351 bp的核苷酸序列,唯有第1个Exon差别较大,但也仅相差15 bp。而对于Intron区域所包含的长度,这2个基因均有差异,即有2~297 bp的长度差异。在此基础上,对这2个物种CesA4基因对应的每一Exon和Intron的同源性进行了比较(表 1)。由表 1可见,PtCesA4与AtCesA4基因内部每一Exon和Intron的同源性均不相同,Exon的同源性由大到小的顺序为Exon 7(84.8%)>Exon 12(79.6%)>Exon 11(78.3%)>Exon 9(77.0%)> Exon 6(74.4%)>Exon 8(71.4%)> Exon 4(70.1%)> Exon 10(69.0%)>Exon 2(67.9%)> Exon 5(66.2%)>Exon 1(38.9%),而Intron的同源性为34.8%~52.2%。这一结果显示,CesA4基因在进化过程中,基因每一部分所受的选择压不同,与CesA4基因的Intron区域相比,Exon区域则较为保守,特别是Exon 6—Exon 9和Exon 11—Exon 12相当保守,同源性均高于70%。
|
|
通过Internet进入NCBI,PtCesA4基因序列的NCBI检索结果初步表明PtCesA4是毛白杨中首次克隆的纤维素合酶CesA4基因。为了分析PtCesA4基因蛋白质的一级结构特征,将PtCesA4 cDNA推导的氨基酸序列与拟南芥AtCesA4、水稻OsCesA1、火炬松(Pinus taeda)PtaCesA2进行了同源比较和结构分析(图 2)。由图 2可见,PtCesA4与拟南芥AtCesA4、水稻OsCesA1和火炬松的PtCesA2蛋白质氨基酸序列高度同源,同源性分别为80.3%,78.9%和75.6%。由图 2显示的CesA蛋白质一级结构可见,无论是裸子植物还是被子植物,双子叶植物还是单子叶植物,草本还是木本植物的CesA的同源性高与低,它们都含有植物纤维素合酶CesA所具有的相对保守的结构域,如在N端存在1个锌指状结构域,位于PtCesA4蛋白质氨基酸序列的第31—76区域,在该结构域中具有4个非常保守的串联重复的半胱氨酸残基序列CxxC,此序列可与DNA结合,推测其与CesA蛋白各亚基之间的相互作用有关(Joshi et al., 2004)。在其蛋白质氨基酸序列内还含有8个高度保守的跨膜结构域,2个在N端(分别位于PtCesA4氨基酸序列的217—238和250—267区域),6个在C端(分别位于PtCesA4氨基酸序列的814—841、851—871、888—913、941—960、970—991和1004—1023区域),其主要功能被认为是在纤维素生物合成过程中,参与了蛋白质间的互作(Samuga et al., 2004)。在PtCesA4氨基酸序列的第268—590和718—813区域分别存在2个亚结构域(Subdomains)A和B,被认为与纤维素合酶的糖苷转移催化活性有关;此外,在Subdomains A和B内含有糖苷转移酶特有的3个保守的D残基(位于342,510和743)和QVLRW区域,被认为与β-糖苷转移酶对底物的结合和催化有关(Joshi et al., 2004)。因此,从PtCesA4蛋白质显示的一级结构可初步看出,与模式植物如拟南芥和水稻一样,PtCesA4也具有植物CesA所具有的结构域和序列特征。

|
图 2 PtCesA4与其他植物CesA蛋白质一级结构比较 Figure 2 Comparison of the primary structure of PtCesA4 with CesAs from different plants PtCesA4:毛白杨CesA4蛋白CesA4 protein of P.tomentosa;AtCesA4:拟南芥CesA4蛋白CesA4 protein of A.thaliana;OsCesA1:水稻CesA1蛋白CesA1 protein of O.sativa;PtaCesA2:火炬松CesA2蛋白CesA2 protein of P.taeda;Zinc-binding domain:锌指状结构域;TMD:转膜结构域Transmembrane domain. |
为了分析毛白杨PtCesA4与其他植物CesA蛋白的系统发育进化关系,利用ClustalX软件(Thompson et al., 1997)将在NCBI注册的拟南芥、玉米、大麦、巨桉(Eucalyptus grandis)、美洲山杨与火炬松等8个物种共31个CesA基因成员的蛋白氨基酸序列进行了序列排列,后利用Treeview软件(Page,1996)得到了这些CesA蛋白的系统进化树(图 3)。有研究者从起源于28亿年前、能够进行光合作用产生多聚糖的螺旋藻(Cyanobacteria)体内检测到有CesA合成纤维素这一现象可以推测,微管植物CesA的起源相当古老(Nobles et al., 2001)。由图 3建立的植物CesA发育进化树可见,植物CesA蛋白由极点向3个方向发生了进化,即形成了3个分支。在第1分支中又分为3个亚分支,其中又分别向双子叶植物和单子叶植物,或裸子植物、被子植物(双子叶植物和单子叶植物)和木本与草本植物方向发生分歧。在第2分支中,进化一开始就分为2个亚分支,其中AtCesA8单独成为一亚分支,而另一亚分支则向裸子植物如火炬松PtaCesA2和被子植物发生分歧,在向被子植物这一进化过程中,又分为单子叶植物如水稻OsCesA1和玉米ZmCesA10和双子叶植物如1个拟南芥CesA蛋白成员和3个树木CesA蛋白成员。在双子叶植物方向包含的这4个CesA蛋白成员中,又向草本如拟南芥AtCesA4和木本方向发生分歧,而在木本方向这一亚分支中,又朝向桉树和杨树这2个物种方向发生进化,其中PtCesA4就是本文克隆的毛白杨CesA4基因蛋白。在第3分支中,分别向2个亚分支方向发生进化,一亚分支包含拟南芥的4个CesA成员(AtCesA2,AtCesA9,AtCesA5和AtCesA6),而另一亚分支又分别向木本(EgCesA6和PtrCesA6)和草本方向(HvCesA2,ZmCesA7,ZmCesA6和ZmCesA8)发生进化分歧。由图 3显示的CesA基因蛋白进化树可见,CesA基因蛋白成员的分化有些出现在物种分化之前(3个分支中的CesA成员),而有些则出现在物种分化之后(亚分支中的某些CesA成员)。

|
图 3 PtCesA4蛋白与其他植物CesA的系统发育进化树 Figure 3 The phylogenetic tree of PtCesA4 with CesAs of the other plants At:拟南芥Arabidopsis thaliana;Os:水稻Oryza sativa;Zm:玉米Zea mays;Eg:巨桉Eucalyptus grandis;Pta:火炬松Pinus taeda;Ptr:毛果杨Populus trichocarpa;Pt:毛白杨Populus tomentosa;Hv:大麦Hordeum vulgare. |
依据PtCesA4基因的mRNA序列,设计1对能够扩增出62 bp左右的cDNA片段的引物(PtCesA4 RTF和PtCesA4 RTR),以逆转录合成的cDNA第1链为模板,以Actin基因的扩增量作为内参,进行Realtime-PCR扩增。PtCesA4基因在不同组织和器官中的扩增量与内参Actin基因扩增量的比值,即相对表达量如图 4所示。由图 4可见,PtCesA4基因在杨树根、茎、叶片和顶端分生组织中均有表达,但其表达模式却不同:PtCesA4在成熟叶片中表达丰度最高(0.851 3),而在顶端分生组织和根部中表达丰度较低(分别为0.1092和0.1247)。在杨树主干不同组织中表达量差别很大,在未成熟木质部中表达丰度最高(0.349),其次为在成熟木质部中的表达丰度(0.3023),而在树皮和韧皮部中表达量较低,分别为0.08376和0.03872,而在树干形成层中表达量最低,为0.004789。因此,由PtCesA4基因在杨树主干中的表达模式可以看出,其在次生微管组织如未成熟木质部和成熟木质部中的表达量约为在初生微管组织如形成层中的63~73倍。先前在模式植物拟南芥AtCesA4(与杨树PtCesA4相对应)的组织特异性差异表达显示,AtCesA4在拟南芥次生细胞壁加厚过程中高丰度表达(Holland et al., 2000)。而最近在另一重要用材树种巨桉(Eucalyptus grandis)中克隆的EgCesA2的表达模式也初步表明了EgCesA2在桉树次生细胞壁的木质部细胞中特异表达(Lu et al., 2008)。这些研究结果都表明了植物CesA4同源基因参与了次生木质部的发育过程和次生细胞壁的加厚,显示了该基因在木质纤维木材形成过程中的重要调控作用。

|
图 4 PtCesA4基因的组织表达谱分析 Figure 4 Relative transcript abundance of PtCesA4 in different tissues and organs R:根Root;B:树干表皮Bark of stem;P:树干韧皮部Phloem of stem;C:树干形成层Cambium of stem;IX:树干未成熟木质部Immature xylem of stem;MX:树干成熟木质部Mature xylem of stem;ML:成熟叶Mature leaf;A:顶端分生组织Apical shoot meristem. |
对PtCesA4基因进行群体遗传学和基于SNP的联合遗传学研究的基础是获知PtCesA4基因在毛白杨种内进化史上所发生的单核苷酸突变。为此,在分析其cDNA序列的基础上,以取自毛白杨自然分布区中3个群体,包含9个省、市、自治区,能够最大范围覆盖毛白杨生长区域的40株基因型个体为材料,对包含297 bp的5′未翻译区域(5′un-translated region,5′UTR)、3 129 bp的外显子、1 553 bp的内含子和331 bp的3′UTR等共5 310 bp的核苷酸序列进行了单核苷酸多态性分析(表 2)。由表 2可见,在PtCesA4基因的5′UTR、外显子、内含子和3′UTR分别检测到了13,69,63和8个SNPs,其出现频率分别为1/23 bp、1/45 bp、1/25 bp和1/41 bp。因此,在PtCesA4基因内部共检测到153个SNPs,SNP出现的频率为1/35 bp(表 2)。由这一结果可以推测,对于PtCesA4基因,在其基因内部的不同区域,其核苷酸变异程度不同,即不同区域的保守性不同,在PtCesA4基因编码区域,其核苷酸变异程度最低,显示了PtCesA4基因在进化过程中编码区域受到了较强的选择压(Suha et al., 2005)。为了分析毛白杨PtCesA4基因内核苷酸突变的替代类型,对检测到的153个SNPs进行了变异类型统计(表 3)。由表 3可见,在这些SNPs中,有118个属于转换类型,分别包含55个G⇔A和63个C⇔T。对于颠换替代类型,共检测到35个,分别包括8个C⇔A、10个G⇔T、11个A⇔T和6个C⇔G。因此,转换/颠换为3.37(>2.0),造成这一结果可能是,SNP在CG序列上出现最为频繁,而且多是C→T,原因是CG中C即胞嘧啶常为甲基化的、自发地脱氨后即成为胸腺嘧啶(Suha et al., 2005)。
|
|

|
|

为了检测PtCesA4在毛白杨东北、南部和西北群体内核苷酸变异及群体分化程度,利用DnaSP4.50.3软件对3个群体分别进行了多态性位点数、单体型、单核苷酸多样性、Tajima's D*(Tajima,1989)和Fu and Li's D* (Fu et al., 1993)的中性检验(表 4)。由表 4可见,对于检测到的单核苷酸位点数s、SNP多样性指数πtot、πsil、πs和πn均有差异,但差异不显著。利用Tajima's D*和Fu and Li's D*进行的中性检验结果表明,在东北和南部群体内D*均为负值,这显示了在这2个群体内,PtCesA4在进化过程中存在过剩的罕见SNP,如在东北和南部群体内分别检测到了48和39个罕见SNPs,而在西北群体内仅检测到了27个罕见SNPs。而对于3个群体来说,均为dn < ds,即非同义突变与同义突变的比率 < 1,这一结果显示了在毛白杨物种演化过程中,是纯化选择,而不是自然选择对PtCesA4基因内同义SNP位点起了主要的进化驱动力。
|
|

为了检测PtCesA4基因编码区内核苷酸位点的改变是否影响了其编码氨基酸的序列,在分析SNP多样性的基础上进行了氨基酸的多态性分析。分别对PtCesA4基因编码区内的69个SNPs进行了同义突变、错义突变和无义突变分析(表 5)。由表 5可见,在PtCesA4编码区内的69个SNPs中,有59个属于同义突变,它们均位于密码子的第3个核苷酸上,有10个属于错义突变,而没有检测到无义突变。在错义突变中,有6个位于密码子的第1个核苷酸上、有2个位于密码子的第2个核苷酸上和1个位于密码子的第3个核苷酸上。其中,在外显子1中,检测到1个位于第3个密码子的错义突变,其密码子由原来的CAT突变为CAG,导致对应的氨基酸组氨酸(His)突变为谷氨酰胺(Gln);在外显子2中有2个错义突变位点,分别位于密码子的第1和第2个核苷酸位置上,其密码子分别由ATT突变为GTT和TAT突变为TGT,分别导致编码的氨基酸异亮氨酸(Ile)突变为缬氨酸(Val);在外显子3中检测到1个错义突变,密码子由CAT突变为AAT,导致相应氨基酸组氨酸(His)突变为天门冬酰胺(Asn)。在外显子6中检测到了2个错义突变,分别由TCC突变为TAC和ACT突变为GCT,从而导致相应氨基酸丝氨酸(Ser)突变为酪氨酸(Tyr)和苏氨酸(Thr)突变为丙氨酸(Ala);在外显子7,8,10和12各检测到1个错义突变。因此,对于毛白杨PtCesA4基因,在其编码区内部发生的非同义突变与同义突变的比率为0.17(< 1),由这一结果可推测在PtCesA4基因进化过程中,纯化选择对其非同义位点起了非常重要的作用(Fu et al., 1993)。
|
|

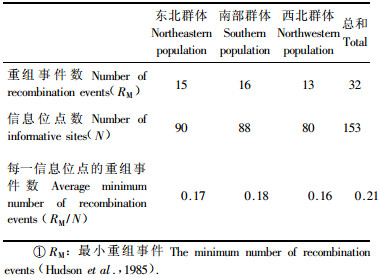
在物种演化过程中,遗传重组是打破LD、提高杂合度和增加遗传多样性的重要驱动力。作者利用Hudson等(1987)建立的方法检测了PtCesA4基因在毛白杨天然分布区的3个群体内SNP间发生的重组事件(表 6)。由表 6可见,在东北、南部和西北群体内检测到的SNP间发生的最小重组事件分别为15,16和13次,将其除以信息位点数后得到每一位点发生的最小重组事件,分别为0.17,0.18和0.16,将3个群体混合后得到总的平均每一信息位点发生的重组事件数为0.21(表 6)。先前在模式植物拟南芥中检测到的SNP间的最小重组事件约为0.08~0.15(Kuittinen et al., 2000),在日本柳杉(Cryptomeria japonica)内检测到的最小重组事件约为0.0000~0.0526(Kado et al., 2003),而在欧洲赤松(Pinus sylvestris)中则为0.25(Dvornyk et al., 2002)。造成这一结果的主要原因可归结为交配系统、有性生殖能力、进化历史和分布范围等因素:如拟南芥是大家熟知的极为容易的自交体系,造成了很低的有效重组率(Kuittinen et al., 2000);而欧洲赤松则为异交体系,分布范围极广,造成了较高的重组率(Dvornyk et al., 2002);毛白杨虽是异交体系、分布广泛,但由于其有性生殖能力低于松树树种,所以其重组率介于拟南芥和欧洲赤松之间。
|
|
理论上,重组增加了基因内部SNP等位位点的多样性,打破了等位位点间的LD,因此,随着基因内部核苷酸长度的延伸,SNP位点间的LD就应该下降。为了理解PtCesA4基因内SNP位点间在毛白杨自然群体内40株基因型个体间LD的延伸长度及程度,利用DnaSP4.50.3软件中的LD程序分析了PtCesA4内SNP的LD水平(图 5)。由图 5可见,随着PtCesA4基因核苷酸序列长度的增加,SNPs的连锁不平衡程度逐渐削弱,当长度达1000 bp左右时,即R2 < 0.1,连锁不平衡迅速消失。这一结果表明,在毛白杨中,SNP的连锁不平衡在候选基因内部就已衰退。而先前进行的人类、动物和植物的LD作图策略中,大体上可分为2类:一是全基因组范围内进行的LD作图,另一则是基于候选基因的LD作图(Zhang et al., 2005)。究竟应采用何种方法进行作图则取决于LD在目标物种基因组中延伸的长度。LD在人类中可从5 kb延伸到500 kb,这使得在人类中进行全基因组范围的LD作图是可行的(Reich et al., 2001)。然而,在植物中LD的延伸范围变异很大,一般来说,在自交物种如在拟南芥中可延伸至250 kb(Nordborg et al., 2002),在大麦中甚至可延伸至10 cM(Kraakman et al., 2004)。相反,在异交物种如玉米中LD延伸仅在1 kb之内就已消失(Remington et al., 2001)。而本研究对毛白杨PtCesA4基因内SNPs的LD水平分析显示,在毛白杨中,基于目标基因的LD作图是可行的,而基于整个杨树基因组的LD作图是不可行的,也是不必要的。

|
图 5 毛白杨PtCesA4基因内SNPs的连锁不平衡 Figure 5 Linkage disequilibrium of SNPs in PtCesA4 |
朱之悌. 2005. 毛白杨遗传改良. 北京: 中国林业出版社, 26-32.
|
Altschul S F, Madden T L, Schaffer A A, et al. 1997. Gapped BLAST and PSI-BLAST:a new generation of protein database search programs. Nucleic Acids Res, 25: 3389-3402. DOI:10.1093/nar/25.17.3389 |
Burton R A, Shirley N J, King B J, et al. 2004. The CesA gene family of barley. Quantitative analysis of transcripts reveals two groups of coexpressed genes. Plant Physiol, 134: 224-236. |
Chiang V L, Puumala R J, Takeuchi H, et al. 1988. Comparison of softwood and hardwood kraft pulping. Tappi J, 71: 173-176. |
Djerbi S, Lindskog M, Arvesrad L. 2005. The genome sequence of black cottonwood (Populus trichocarpa) reveals 18 conserved cellulose synthase (CesA) genes. Planta, 221: 739-746. DOI:10.1007/s00425-005-1498-4 |
Dvornyk V, Sirvio A, Mikkonen M, et al. 2002. Low nucleotide diversity at the pall locus in the widely distributed Pinus sylvestris. Mol Biol Evol, 19: 179-188. DOI:10.1093/oxfordjournals.molbev.a004070 |
Fu Yunxin, Li Wenhsiung. 1993. Statistical tests of neutrality of mutatons. Genetics, 133: 693-709. |
Holland N, Holland D, Helentjaris T, et al. 2000. A comparative analysis of the plant cellulose synthase (CesA) gene family. Plant Physiol, 123: 1313-1323. DOI:10.1104/pp.123.4.1313 |
Hudson R R, Kaplan N L. 1985. Statistical properties of the number of recombination events in the history of a sample of DNA sequences. Genetics, 111: 147-164. |
Hudson R R, Kreitman M, Aguade M. 1987. A tests of neutral molecular evolution based on nucleotide data. Genetics, 116: 153-159. |
Joshi C P, Bhandari S, Ranjan P, et al. 2004. Genomics of cellulose biosynthesis in poplars. New Phytologist, 164: 53-61. DOI:10.1111/j.1469-8137.2004.01155.x |
Kado T, Yoshimaru H, Tsumura Y, et al. 2003. DNA variation in a conifer, Cryptomeria japonica (Copressaceae sensu lato). Genetics, 164: 1547-1599. |
Kimura S, Kondo T. 2002. Recent progress in cellulose biosynthesis. J Plant Res, 115: 297-302. DOI:10.1007/s10265-002-0037-7 |
Kraakman A T, Niks W R E, Van den Berg P M M M, et al. 2004. Linkage disequilibrium mapping of yield and yield stability in modern spring barley cultivars. Genetics, 168: 435-446. DOI:10.1534/genetics.104.026831 |
Kuittinen H, Aguage M. 2000. Nucleotide variation at the chalcone isomerase locus in Arabidopsis thaliana. Genetics, 155: 863-872. |
Lu Shanfa, Li Laigeng, Yi Xiaoping. 2008. Differential expression of three eucalyptus secondary cell wall-related cellulose synthase genes in response to tension stress. Journal of Experimental Botany, 59: 681-695. DOI:10.1093/jxb/erm350 |
Nobles D R, Romanovicz D K, Brown Jr R M. 2001. Cellulose in Cyanobacteria Origin of vascular plant cellulose synthase?. Plant Physiol, 127: 529-542. DOI:10.1104/pp.010557 |
Nordborg M, Borevitz J O, Bergelson J, et al. 2002. The extent of linkage disequilibrium in Arabidopsis thaliana. Nat Genet, 30: 190-193. DOI:10.1038/ng813 |
Page R D. 1996. TreeView:an application to display phylogenetic trees on personal computers. Comput Appl Biosci, 12: 357-358. |
Pear J R, Kawagoe Y, Schreckengost W E, et al. 1996. Higher plants contain homologs of the bacterial celA genes encoding the catalytic subunit of cellulose synthase. PNAS, 93: 12637-12642. DOI:10.1073/pnas.93.22.12637 |
Reich D E, Cargill M, Bolk S, et al. 2001. Linkage disequilibrium in the human genome. Nature, 411: 199-204. DOI:10.1038/35075590 |
Remington D L, Thornsberry J M, Matsuoka Y, et al. 2001. Structure of linkage disequilibrium and phenotypic associations in the maize genome. PNAS, 98: 11479-11484. DOI:10.1073/pnas.201394398 |
Richmond T A, Somerville C R. 2000. The cellulose synthase superfamily. Plant Physiol, 124: 495-498. DOI:10.1104/pp.124.2.495 |
Samuga A, Joshi C P. 2004. Cloning and characterization of cellulose synthase-like gene, PtrCSLD2 from developing xylem of aspen trees. Physiologia Plantarum, 120: 631-641. DOI:10.1111/ppl.2004.120.issue-4 |
Sassner P, Galbe M, Zacchi G. 2005. Steam pretreatment of Salix with and without SO2 impregnation for production of bioethanol. Appl Biochem Biotech, 124: 1101-1118. DOI:10.1385/ABAB:124:1-3 |
Saxena I M, Lin F C, Brown R M, et al. 1990. Cloning and sequencing of the celluose synthase catalytic subunit gene of Acetobacter xylinum. Plant Mol Biol, 15: 673-683. DOI:10.1007/BF00016118 |
Saxena I M, Brown R M. 1995. Identification of a second cellulose synthase gene (acsAII) in Acetobacter xylinu. J Bacteriol, 177: 5276-5283. DOI:10.1128/jb.177.18.5276-5283.1995 |
Sterky F, Bhalerao R R, Unneberg P, et al. 2004. A Populus EST resource for plant functional genomics. PNAS, 101: 13951-13956. DOI:10.1073/pnas.0401641101 |
Suha Y, Vijg J. 2005. SNP discovery in associating genetic variation with human disease phenotypes. Mutation Research, 573: 41-53. DOI:10.1016/j.mrfmmm.2005.01.005 |
Tajima F. 1989. Statistical method for testing the neutral mutation hypothesis by DANN polymorphism. Genetics, 123: 585-595. |
Tanaka K, Murata K, Yamazaki M, et al. 2003. Three distinct rice cellulose synthase catalytic subunit genes required for cellulose synthesis in the secondary wall. Plant Physiol, 133: 73-83. DOI:10.1104/pp.103.022442 |
Thompson J D, Gibson T J, Plewniak F, et al. 1997. The CLUSTAL X windows interface:flexible strategies for multiple sequence alignment aided by quality analysis tools. Nucleic Acids Res, 25: 4876-4882. DOI:10.1093/nar/25.24.4876 |
Wu Luguang, Joshi C P, Chiang V L. 2000. A xylem-specific cellulose synthase gene from aspen (Populus tremuloides) is responsive to mechanical stress. Plant J, 22: 495-502. DOI:10.1046/j.1365-313x.2000.00758.x |
Wullschleger S D, Jansson S. 2002. Genomics and forest biology:Populus emerges as the perennial favorite. The Plant Cell, 14: 2651-2655. DOI:10.1105/tpc.141120 |
Zhang Deqiang, Zhang Zhiyi. 2005. Single nucleotide polymorphisms discovery and linkage disequilibrium. Forestry Studies in China, 7: 1-14. |