 2009, Vol. 45
2009, Vol. 45文章信息
- 杨晓慧, 张有慧, 张志毅, 李百炼, 张德强.
- Yang Xiaohui, Zhang Youhui, Zhang Zhiyi, Li Bailian, Zhang Deqiang
- 毛白杨干细胞决定基因Wuschel的克隆及其单核苷酸多态性分析
- Isolation and Single Nucleotide Polymorphisms Analysis of Stem Cell Organizer Gene Wuschel in Populus tomentosa
- 林业科学, 2009, 45(1): 43-49.
- Scientia Silvae Sinicae, 2009, 45(1): 43-49.
-
文章历史
- 收稿日期:2008-10-08
-
作者相关文章



2. 山东省国营冠县苗圃 冠县 252525;
3. 美国北卡罗莱纳州立大学林学系 北卡罗莱纳州 NC27695-8203
2. Guanxian County Tree Nursery of Shandong Province Guanxian 252525;
3. Department of Forestry, North Carolina State University North Carolina State 27695-8203
植物干细胞位于茎或根的顶端分生组织,是植物胚后发育过程中新器官产生和组织发育的源泉。与动物干细胞一样,植物干细胞具有无限增殖和分化为多种细胞类型的能力(Byrne et al., 2003)。在具有次生生长的植物中,顶端分生组织(shoot apical meristem, SAM)可有序地产生原形成层(procambium)和微管形成层(vascular cambium),并进一步分化为初生维管组织(初生木质部和初生韧皮部)和次生维管组织(次生木质部和次生韧皮部)。而对于林木来说,森林的主要产品——次生木质部就是向社会提供的生物质纤维原料。因此,植物干细胞,特别是树木主干微管形成层干细胞在树木生长和木材形成过程中具有极其重要的生物学意义。
对模式植物如拟南芥(Arabidopsis thaliana)和主要经济树种如杨树、桉树和松树木材形成的基因组学研究表明,植物微管系统发育在进化上相对保守,木材微管形成层和次生木质部的发育并非树木所独有(Baucher et al., 2007)。因此,分子遗传学家建议:对多年生树木形成层和次生生长的研究可以借鉴一年生模式植物木材形成机制方面的研究成果,以此来推动木本植物该方面的研究进程。另一方面,随着基因组学和现代分子生物学工具如基因芯片技术在树木微管组织形成和发育中的广泛应用,特别是杨树全基因组测序工作的完成和许多可利用的表达标签(expressed sequence tags, ESTs)数据库的建立,为研究杨树木材形成过程中干细胞维持和分化的基因调控机制提供了可能(Plomion et al., 2005; Tuskan et al., 2006)。
植物Homeobox转录因子基因Wuschel(Wus)的编码产物是维持干细胞数量的内源性信号分子。Wus基因首先在胚胎形成的早期阶段表达,然后迅速转向仅在SAM干细胞组织中心区域的内层部分表达(Schoof et al., 2000)。对拟南芥Wus突变体的研究发现,由于Wus基因的突变导致分裂组织的终止和干细胞功能的丧失,如花的数量明显减少且提前脱落等(Laux et al., 1996)。而Wus基因的超量表达致使SAM中心区域干细胞大量增殖(Schoof et al., 2000)。由此可见,Wus基因主要是通过维持SAM中心区域干细胞群的数量和特异性来实现提高茎和花的分生组织活性这一功能。而Wus基因的异位表达则诱导异位干细胞的形成,表明Wus表达区域必须受到严格的调控以维持干细胞的正确位置和数量(Mayer et al., 1998)。这些研究初步表明,在植物器官和组织初生和次生生长过程中,Wus基因在干细胞维持和分化的调控途径中具有极其重要的作用(Fletcher, 2002; Byrne et al., 2003; Sharma et al., 2003; Morrison et al., 2008)。
Wus基因成员虽已从拟南芥、水稻(Oryza sativa)、玉米(Zea mays)中分离出来并进行了功能性检测,但对于高度杂合、世代周期较长、树体高大,对经济建设和环境保护具有重要作用的用材树种来说,更有必要对该基因进行分离和检测。为此,本研究以拟南芥干细胞决定基因Wus的氨基酸序列为信息探针, 基于可利用的杨树EST数据库和毛果杨(Populus trichocarpa)全基因组测序结果,通过电子杂交辅助的克隆技术,从毛白杨(Populus tomentosa)形成层cDNA中分离得到2个Wus基因成员。在此基础上,进行了这2个候选基因的单核苷酸多态性(single nucleotide polymorphisms, SNPs)和连锁不平衡分析。本研究为分离其他树种Wus成员以及进行该基因的连锁不平衡作图和基因辅助树木育种提供了重要的理论依据。
1 材料与方法 1.1 植物材料1983—1984年,全国毛白杨协作组从毛白杨分布区如北京、河北、山东、河南、山西、陕西、甘肃、宁夏、安徽和江苏等10个省(市、自治区)的100个种源中,选取了1 047株基因型个体,通过根繁方式在山东省冠县国营苗圃建立了全国毛白杨基因库。本研究提取DNA的叶片材料取自该基因库中能够最大程度地反映毛白杨分布范围的36个种源,即36株基因型个体,并置于液氮中冻存备用。
提取RNA的材料取自中国林业科学研究院内毛白杨雌株的形成层组织,置于液氮中冻存备用。
1.2 总DNA的提取按Murray和Thompson(1980)描述的方法进行。
1.3 RNA提取和cDNA的合成毛白杨茎形成层总RNA的提取和cDNA的合成分别按RNeasy Plant Mini Kits (Qiagen, Inc., Valencia, CA, USA)和Clonetech试剂盒描述的方法进行。
1.4 电子杂交以拟南芥AtWus的氨基酸序列(GenBank注册号为ATH012310)为信息探针, 将其输入瑞典和法国杨树EST数据库(Sterky et al., 2004)以及毛果杨全基因组数据库(Wullschleger et al., 2002)同源比较服务器进行BLASTX分析(Altschul et al., 1997), 筛选出Wus蛋白质结构域同源性在75%以上的ESTs和Contigs作为候选序列, 然后逐一将这些序列送GenBank核酸数据库检索, 后将这些序列进行整合拼接, 获得统计上完整的序列。
1.5 电子杂交探针制备根据所得的EST重叠群整合序列, 在其可能的开放阅读框两端设计一对寡聚核苷酸引物:
PtWus1F: 5′-CTCAATCCATCATGGAACCT-3′,
PtWus1R: 5′-TGCCCAGCAAGAAGACACC-3′;
PtWus2F: 5′-CTCAATCCATCATGGAACCTCA-3′,
PtWus2R: 5′-TGCCCAGCAAGAAGACACCTTA-3′。
应用25 μL DNA聚合酶链反应(PCR)体系, 以毛白杨形成层cDNA为模板, 加入2.5 μL 10 × buffer, 1.8 μL 25 mmol·L-1 MgCl2, 1 μL 10 mmol·L-1 dNTP,Taq DNA聚合酶1.0 U(以上试剂购自Promega公司), 100 nmol·L-1正向和反向引物各1 μL, 加适量双蒸水至25 μL。于94 ℃, 3 min →(93 ℃, 30 s →54 ℃, 30 s→ 72℃, 1 min) 35个循环→72 ℃, 5 min热循环条件下, 扩增出长约950 bp的cDNA片段。
1.6 PCR产物的克隆、测序及计算机分析将PCR扩增得到的目的基因片段回收后连接于pGEM-T上。连接产物转化大肠杆菌DH5α,筛选阳性克隆,送公司测序。应用DNAMAN6.0软件和CLUSTALX软件包程序推导出氨基酸序列并进行NCBI检索分析, 然后分别估算和预测推导蛋白质的分子质量和等电点。
1.7 Wus基因的SNP检测依据已克隆的2个Wus基因的核苷酸序列设计基因特异的引物,以选取的36株个体的总DNA为模板进行PCR扩增;将PCR扩增产物进行琼脂糖凝胶电泳分离,回收、纯化目的片段后与PGEM-T载体连接,转化后挑取阳性单克隆进行序列测定,然后将每一基因片段的核苷酸序列拼接成完整的基因序列。
1.8 Wus基因的SNP多样性分析和连锁不平衡检测利用MEGA3.0软件对每一基因的36个序列进行分析,标出SNP位点,计算SNP频率、转换和颠换的SNP数量;计算SNP多样性指数、在基因不同区域的分布模式;分析同义突变、错义突变和无义突变以及发生在剪切位点(GT-AG)的突变情况;分析LD在不同基因中的延伸模式。
2 结果与讨论 2.1 毛白杨Wus同源cDNA的克隆及其结构特征分析对于Wus基因,利用电子杂交的策略在毛果杨全基因组上共获得了2个Wus候选序列,分别命名为PtWus1和PtWus2,根据理论序列, 设计了能扩增出编码区全长的2对引物PtWus1F和PtWus1R以及PtWus2F和PtWus2R, 以毛白杨形成层总cDNA为模板进行PCR扩增。扩增产物经0.8%琼脂糖凝胶电泳分离(图 1),在大小约为950 bp处各扩增出一条明亮的特异条带。将扩增的特异片段回收、纯化并与pGEM-T载体连接后克隆测序。测序结果表明,克隆的这2个毛白杨Wus cDNA总长分别为922 bp和956 bp,它们的同源性为84.5%。在PtWus1和PtWus2序列中, 基因内部含有完整的开放阅读框架,大小分别为777 bp和795 bp,可编码长度分别为258和264个氨基酸残基的蛋白质(基因注册号分别为FJ232063和FJ232064)。运用DNAMAN6.0软件估算推导的这2个蛋白质的分子质量分别约为29.4 ku和29.5 ku, 其等电点分别为6.78和6.02。

|
图 1 从毛白杨形成层cDNA中扩增的PtWus1和PtWus2cDNA电泳图 Figure 1 The cDNA fragment of PtWus1 and PtWus2 produced by RT-PCR in P. tomentosa M:1 kb DNA ladder; 1:从毛白杨形成层cDNA中扩增出的PtWus1片段The cDNA fragment of PtWus1 amplified from cambium cDNA in P. tomentosa; 2:从毛白杨形成层cDNA中扩增出的PtWus2片段The cDNA fragment of PtWus2 amplified from cambium cDNA in P. tomentosa. |
将PtWus1和PtWus2基因序列与已公开的拟南芥AtWus1基因的homeodomain序列进行同源比较,发现它们与拟南芥AtWus1的同源性分别为76.0%和74.9%。NCBI检索结果初步表明,PtWus1和PtWus2是林木中首次克隆的Wus转录因子基因。为了分析PtWus1和PtWus2基因蛋白质的一级结构特征,将PtWus1和PtWus2cDNA推导的氨基酸序列与拟南芥AtWus1、水稻OsWus1以及玉米ZmWus11和ZmWus2的homeodomain进行了同源比较和结构分析(图 2)。PtWus1与AtWus1、OsWus1和ZmWus1基因的蛋白质氨基酸序列同源,同源性分别为77.0%、65.1%和77.0%,而PtWus2与AtWus1、OsWus1和ZmWus2的同源性分别为77.0%、65.1%和73.8%,PtWus1和PtWus2的氨基酸同源性为80.4%。由图 2显示的Wus基因的蛋白质一级结构可见,无论PtWus1和PtWus2之间及其与拟南芥、水稻和玉米Wus之间的同源性高与低,它们都含有高度保守的homeodomain区域,分别位于PtWus1氨基酸序列的第33—93氨基酸之间和PtWus2氨基酸序列的第27—87氨基酸之间(Mayer et al., 1998)。

|
图 2 PtWus1和PtWus2与其他植物Wus蛋白质一级结构比较 Figure 2 Comparison of the primary structure of Wus sequences from different plants PtWus1:毛白杨Wus1蛋白Wus1 protein of Populus tomentosa;PtWus2:毛白杨Wus2蛋白Wus2 protein of P. tomentosa;AtWus1:拟南芥Wus1蛋白Wus1 protein of Arabidopsis thaliana;OsWus1:水稻Wus1蛋白Wus1 protein of Oryza sativa;ZmWus1:玉米Wus1蛋白Wus1 protein of Zea mays;ZmWus2:玉米Wus2蛋白Wus2 protein of Z. mays. |
为了分析杨树PtWus1和PtWus2与其他植物Wus蛋白的系统发育进化关系,利用ClustalX软件(Thompson et al., 1997)将杨树PtWus1和PtWus2及在NCBI注册的拟南芥AtWus1、玉米ZmWus1与ZmWus2和水稻OsWus1 4个物种共6个Wus基因的蛋白氨基酸序列进行了序列排列,后利用Treeview软件(Page, 1996)得到了这些Wus蛋白的系统进化树(图 3)。可以看出,Wus基因蛋白由极点向2个方向发生了进化,分为双子叶植物和单子叶植物。在双子叶植物这一分支中,包括AtWus1、PtWus1和PtWus2,这一分支又分为2个亚分支,其中一亚分支又分为PtWus1和PtWus2,即是本文分离的2个毛白杨Wus转录因子基因。由图 3显示的Wus基因蛋白进化树可见,Wus基因蛋白的亚家族成员如PtWus1和PtWus2,以及ZmWus1与ZmWus2的分化出现在双子叶植物和单子叶植物物种分化之后。

|
图 3 PtWus1和PtWus2与其他植物Wus的系统发育进化树 Figure 3 The phylogenetic tree of PtWus1 and PtWus2 with Wus of the other plants |
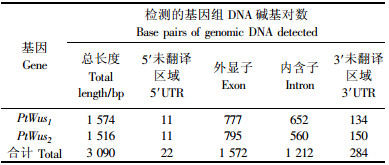
在分析其cDNA序列的基础上,对毛白杨PtWus1和PtWus2基因进行了基因组结构分析,测序结果表明,克隆的毛白杨PtWus1和PtWus2基因的基因组DNA长度分别为1 574 bp和1 516 bp(表 1),其中均包含11 bp的5′未翻译区域(5′un-translated region, 5′UTR),分别包含134 bp和150 bp的3′UTR,且分别包含777 bp和795 bp的编码区域。与模式植物拟南芥一样,毛白杨PtWus1和PtWus2基因的DNA序列也含有2个内含子,对于PtWus1,内含子长度分别为142 bp与510 bp,而对于PtWus2,内含子则分别为106 bp与454 bp。因此,对于杨树Wus蛋白基因,共对其基因组3 090个核苷酸序列进行了单核苷酸多态性分析,其中包括22 bp的5′UTR、1 572 bp的外显子、1 212 bp的内含子和284 bp的3′UTR(表 1)。
|
|
在DNA序列分析的基础上,对PtWus1和PtWus2基因分别进行了SNP多样性分析(表 2)。由表 2可见,在PtWus1和PtWus2基因内部分别检测到58个和51个SNPs,SNP的多样性分别为1/27 bp和1/30 bp。对于PtWus1,在检测到的58个SNPs中,24个属于常见SNPs(在群体中出现频率超过10%),而34个属于罕见SNPs。在这些SNPs中,有10、10、3、22、8和5个分别位于PtWus1基因的外显子1、内含子1、外显子2、内含子2、外显子3和3′ UTR区域(表 2)。而对于PtWus2,在检测到的51个SNPs中,有28个常见SNPs,23个罕见SNPs。在这些SNPs中,有7、6、2、15、17和4个分别位于PtWus2基因的外显子1、内含子1、外显子2、内含子2、外显子3和3′UTR区域(表 2)。因此,对于杨树这2个Wus蛋白基因,共检测到了109个SNPs,SNP的多样性为1/28 bp,除5′UTR,由于序列太短,没有检测到单核苷酸突变外,SNP在外显子、内含子和3′ UTR的多样性差异较大,分别为1/33 bp、1/23 bp和1/31 bp。由这一结果可以推测,对于Wus蛋白基因,在其基因内部的不同区域,其核苷酸变异程度不同,即不同区域的保守性不同,这主要是由于在进化过程中受到的选择压不同所致。
|
|

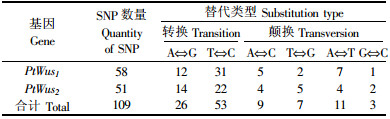
为了分析杨树Wus基因的单核苷酸突变替代类型,对检测到的109个SNPs进行了变异类型统计(表 3)。由表 3可见,在这些SNPs中,有79个属于转换类型,分别包含26个G⇔A和53个C⇔T。对于颠换替代类型,共检测到30个,分别包括9个C⇔A、7个G⇔T、11个A⇔T和3个C⇔G。其中,对于PtWus1,有43个SNPs属于转换,分别包含12个G⇔A和31个C⇔T;而有15个属于颠换,分别包含5个C⇔A、2个G⇔T、7个A⇔T和1个C⇔G。而对于PtWus2,共有36个属于转换,分别包含14个G⇔A和22个C⇔T;有15个属于颠换,分别包含4个C⇔A、5个G⇔T、4个A⇔T和2个C⇔G。PtWus1、PtWus2的转换/颠换分别约为2.9和2.4,均超过2.0,这一结果与Suha和Vijg(2005)对人类候选基因的单核苷酸替代类型的分析结果相一致。
|
|
为了检测PtWus1和PtWus2基因编码区内核苷酸位点的改变是否影响了其编码氨基酸的序列,在分析SNP多样性的基础上进行了氨基酸的多态性分析。分别对PtWus1和PtWus2基因编码区内的21和26个SNPs进行了同义突变、错义突变和无义突变分析(表 4)。由表 4可见,在PtWus1编码区内的21个SNPs中,16个属于同义突变,它们均位于密码子的第3个核苷酸上,4个属于错义突变,1个属于无义突变。在错义突变中,有2个位于外显子1中,其密码子分别由原来的CAA突变为CAT,以及由ACT突变为ATT,从而导致对应的氨基酸谷氨酰胺(Gln)变为组氨酸(His),以及苏氨酸(Thr)变为异亮氨酸(Ile);有1个位于外显子2中,其密码子由AAT突变为ACT,导致编码的氨基酸天门冬酰胺(Asn)变为苏氨酸(Thr);有1个位于外显子3中,密码子由ATT突变为ATG,导致相应氨基酸异亮氨酸(Ile)变为起始密码子(Met)(表 4)。在PtWus1中,同义突变、错义突变和无义突变的频率分别为76%、19%和5%,同义突变与非同义突变的比率为3.2。而对于PtWus2,在编码区内检测到的26个SNPs中,同义突变和错义突变均为13个,其突变频率也均为50%,同义突变与非同义突变的比率为1.0;在13个错义突变中,有11个位于密码子的第1和第2的核苷酸位置上,它们的改变均导致了氨基酸序列的变化,有2个位于密码子的第3个核苷酸上。因此,对于毛白杨Wus基因,在其编码区内部共检测到了29个同义突变和18个非同义突变,非同义突变与同义突变的比率为0.6(< 1),特别是PtWus1基因,其非同义突变/同义突变仅为0.3,这一结果初步表明在PtWus1基因的进化过程中,纯化选择起了非常重要的作用。
|
|

连锁不平衡(linkage disequilibrium, LD)在目标物种基因组中延伸的长度决定了LD作图的方式。LD在人类中可从5 kb延伸到500 kb,这使得在人类中进行全基因组范围的LD作图是可行的(Reich et al., 2001)。然而,在植物中LD的延伸范围变异很大,一般来说,在自交物种如在拟南芥中可延伸至250 kb(Nordborg et al., 2002),在大麦(Hordeum vulgare)中甚至可延伸至10 cM(Kraakman et al., 2004)。相反,在异交物种如玉米中LD延伸仅在1 kb之内就已消失(Remington et al., 2001)。
为了检测毛白杨PtWus1和PtWus2基因内部SNP位点的LD情况,利用DnaSP4.0软件对毛白杨PtWus1和PtWus2基因在36株基因型个体中的连锁不平衡情况进行了分析(图 4)。由图 4可见,随着PtWus1和PtWus2基因核苷酸序列长度的增加,SNPs的连锁不平衡程度逐渐消弱,但长度达750 bp左右时,即R2 < 0.3,连锁不平衡迅速消失。这一结果表明,在毛白杨中,SNP的连锁不平衡在候选基因内部就已衰退。因此,在毛白杨中,基于候选基因的连锁不平衡作图是可行的,基于整个杨树基因组的连锁不平衡作图是不可行的,也是不必要的。

|
图 4 毛白杨PtWus1和PtWus2基因内SNPs的连锁不平衡 Figure 4 Linkage disequilibrium(LD) of SNPs in PtWus1 and PtWus2 in P. tomentosa |
Altschul S F, Madden T L, Schaffer A A, et al. 1997. Gapped BLAST and PSI-BLAST: a new generation of protein database search programs. Nucleic Acids Res, 25: 3389-3402. DOI:10.1093/nar/25.17.3389 |
Baucher M, Jaziri M E, Vandeputte O. 2007. From primary to secondary growth: origin and development of the vascular system. Journal of Experimental Botany, 58: 3485-3501. DOI:10.1093/jxb/erm185 |
Byrne M E, Kidner C A, Martienssen R A. 2003. Plant stem cells: divergent pathways and common themes in shoots and roots. Current Opinion in Genetics and Development, 13: 551-557. DOI:10.1016/j.gde.2003.08.008 |
Fletcher J C. 2002. Coordination of cell proliferation and cell fate decisions in the angiosperm shoot apical meristem. BioEssays, 24: 27-37. DOI:10.1002/(ISSN)1521-1878 |
Kraakman A T, Niks W R E, Van den Berg P M M M, et al. 2004. Linkage disequilibrium mapping of yield and yield stability in modern spring barley cultivars. Genetics, 168: 435-446. DOI:10.1534/genetics.104.026831 |
Laux T, Mayer K F X, Berger J, et al. 1996. The WUSCHEL gene is required for shoot and floral meristem integrity in Arabidopsis. Development, 122: 87-96. |
Mayer K F X, Schoof H, Haecker A, et al. 1998. Role of WUSCHEL in regulating stem cell fate in the Arabidopsis shoot meristem. Cell, 95: 805-815. DOI:10.1016/S0092-8674(00)81703-1 |
Morrison S J, Spradling A C. 2008. Stem cells and niches: mechanisms that promote stem cell maintenance throughout life. Cell, 132: 598-611. DOI:10.1016/j.cell.2008.01.038 |
Murray M G, Thompson W F. 1980. Rapid isolation of high-molecular-weight plant DNA. Nucleic Acids Res, 8: 4321-4325. DOI:10.1093/nar/8.19.4321 |
Nordborg M, Borevitz J O, Bergelson J, et al. 2002. The extent of linkage disequilibrium in Arabidopsis thaliana. Nat Genet, 30: 190-193. DOI:10.1038/ng813 |
Page R D. 1996. TreeView: an application to display phylogenetic trees on personal computers. Comput Appl Biosci, 12: 357-358. |
Plomion C, Richardson T, MacKay J. 2005. Advances in forest tree genomics. New Phytologist, 166: 705-708. DOI:10.1111/j.1469-8137.2005.01445.x |
Reich D E, Cargill M, Bolk S, et al. 2001. Linkage disequilibrium in the human genome. Nature, 411: 199-204. DOI:10.1038/35075590 |
Remington D L, Thornsberry J M, Matsuoka Y, et al. 2001. Structure of linkage disequilibrium and phenotypic associations in the maize genome. PNAS, 98: 11479-11484. DOI:10.1073/pnas.201394398 |
Schoof H, Lenhard M, Haecker A, et al. 2000. The stem cell population of Arabidopsis shoot meristems is maintained by a regulatory loop between the CLAVATA and WUSCHEL genes. Cell, 100: 635-644. DOI:10.1016/S0092-8674(00)80700-X |
Sharma V K, Carles C, Fletcher J C. 2003. Maintenance of stem cell populations in plants. PNAS, 100: 11823-11829. DOI:10.1073/pnas.1834206100 |
Suha Y, Vijg J. 2005. SNP discovery in associating genetic variation with human disease phenotypes. Mutation Research, 573: 41-53. DOI:10.1016/j.mrfmmm.2005.01.005 |
Sterky F, Bhalerao R R, Unneberg P, et al. 2004. A Populus EST resource for plant functional genomics. PNAS, 101: 13951-13956. DOI:10.1073/pnas.0401641101 |
Thompson J D, Gibson T J, Plewniak F, et al. 1997. The CLUSTAL X windows interface: flexible strategies for multiple sequence alignment aided by quality analysis tools. Nucleic Acids Res, 25: 4876-4882. DOI:10.1093/nar/25.24.4876 |
Tuskan G A, DiFazio S, Jansson S, et al. 2006. The genome of black cottonwood, Populus trichocarpa (Torr. & Gray). Science, 313: 1596-1604. DOI:10.1126/science.1128691 |
Wullschleger S D, Jansson S. 2002. Genomics and forest biology: Populus emerges as the perennial favorite. The Plant Cell, 14: 2651-2655. DOI:10.1105/tpc.141120 |