 2008, Vol. 44
2008, Vol. 44文章信息
- 李绍臣, 高福玲, 姜廷波.
- Li Shaochen, Gao Fuling, Jiang Tingbo.
- 基于RAPD标记的白桦遗传连锁群分析
- Analysis of Genetic Linkage Groups on Birch Using RAPD Markers
- 林业科学, 2008, 44(5): 155-159.
- Scientia Silvae Sinicae, 2008, 44(5): 155-159.
-
文章历史
- 收稿日期:2007-04-25
-
作者相关文章



白桦(Betulaplatyphylla)生长速度快,适应性强,材质优良,是重要的工业用材树种,也是伐区、火烧迹地天然更新的先锋树种。自“八五”以来,人们对白桦扦插繁殖(詹亚光等,1994)、种源区划和亲缘关系(朱翔等,2001;姜静等,2001a;2001b)、强化育种(杨传平等,2004)、遗传转化(詹亚光等,2006)、杂交育种(李开隆等,2006)等方面进行了大量研究。白桦和其他树种一样具有生长周期长、遗传改良慢等特点。如何缩短育种周期,在较短的时间培育出符合人们需要的优良白桦新品种是林木育种工作者所面临的问题。分子标记辅助选择是提高林木育种效率的重要手段,明确林木连锁群结构是进行分子育种的理论依据。迄今为止,国内外对白桦连锁群的研究很少,Pekkinen等(2005)报道的欧洲白桦(Betula pendula)遗传图谱是由4个不同家系整合而成,只有101个标记,总连锁群16个,平均图距15.5 cM,图谱总长度1 561 cM,应用价值非常有限。本研究利用拟测交策略(Grattapaglia et al., 1994),采用RAPD分子标记技术,以欧洲白桦×白桦F1代79个个体为研究群体,对欧洲白桦和白桦的遗传连锁群结构进行分析,为实现重要性状的基因定位及分子标记辅助选择育种提供研究基础。
1 材料与方法 1.1 材料亲本选择和人工控制授粉在东北林业大学白桦强化育种园内进行。用RAPD检测育种园优树,选择地理远源、遗传距离较大的欧洲白桦(来自芬兰)为母本,白桦(来自中国帽儿山)为父本。2005年4月中旬进行控制授粉,8月上旬采集成熟果穗,同年10月进行播种育苗,共获得了79株生长发育正常的个体,包括亲本共81株作为作图群体。
1.2 方法1) DNA提取和PCR扩增 以白桦嫩叶为材料,用DNA提取试剂盒(Universal Genomic DNA Extraction Kit Ver.3.0,TaKaRa,大连)分离DNA。PCR扩增反应在PTC-200 PCR仪(MJ research USA)上进行。反应总体积为20 μL:DNA模板50 ng,引物0.5 μmol·L-1,Taq酶(MBI)1.0 U,dNTP各200 μmol·L-1,1×反应缓冲液(10 μmol·L-1 Tris-Cl,pH8.0,50 mmol·L-1 KCl,2.5 mmol·L-1 MgCl2)。扩增程序为:94 ℃预变性4 min;94 ℃变性30 s,36 ℃退火30 s,72 ℃延伸1 min,40个循环;72 ℃延伸7 min。扩增产物在1.0%的琼脂糖凝胶上进行电泳分离和多态性检测分析。
2) 引物筛选 用2个亲本DNA对1 000个寡核苷酸随机引物进行多态性筛选。然后以10个F1个体的DNA为模板,确定在双亲间存在多态性、并且在杂种后代中表现分离的引物。用在双亲和F1中均存在多态性的引物对包括亲本在内的81个个体的DNA进行PCR扩增,扩增产物经电泳检测后,进行数据收集,获得RAPD标记。根据PCR扩增谱带的有无分别对个体标记基因型进行赋值,将某一谱带出现的个体在该位点的标记基因型赋值为ab(杂合型),该谱带不出现的个体在该位点的基因型赋值为aa(纯合型),难于判读的谱带和缺失数据赋值为“-"。
3) 数据分析 利用χ2检验(χ0.05(1)2=3.84)对多态性位点进行检测,确定符合1:1分离的RAPD标记位点用于连锁群分析。采用FslinkageMao 1.0软件(施季森等,2006),在LOD≥3.0,重组率r≤0.5条件下估计连锁群。
4) 连锁图谱的参数计算 遗传图谱实际长度为框架图长度,即连锁群(>3个标记)长度之和;连锁群总长度包括连锁群、三联体和连锁对在内所有连锁长度的总和。参照Chakravarti等(1991)的方法估计遗传图谱长度,即各连锁群实际长度乘(m+1)/(m-1)作为估计长度,m为每个连锁群的标记数。框架图覆盖率等于框架图长度占估算的遗传图谱总长度的百分比;总的图谱覆盖率等于连锁群总长度占估算的遗传图谱总长度的百分比。
2 结果与分析 2.1 RAPD标记在欧洲白桦×白桦F1群体中的分离用178个扩增产物清晰、在双亲中存在多态性、且在子代中有分离的随机寡核苷酸引物对包括双亲在内的81个个体进行随机扩增,检测到296个多态性位点,这些扩增产物的片段长度在500~2 000 bp之间。图 1显示了引物S1066产生的拟测交位点在F1群体中的分离。利用χ2(χ0.05(1)2=3.84)检验出符合拟测交1:1分离的位点276个,其他分离类型的位点23个。在276个符合拟测交分离的位点中,有137个位点来自欧洲白桦,占50.2%;有136个位点来自白桦,占总位点的49.8%。双亲多态性位点在子代中各占50%左右,符合正常杂交组合的位点分布。

|
图 1 引物S1066的扩增片段在欧洲白桦×白桦F1群体中的分离 Figure 1 Segregation of amplified bands by primer S1066 in the B. pendula×B. platyphylla F1 families 1,24:分子量标记Molecular mass marker; 2,25:欧洲白桦B. pendula;3,26:白桦B. platyphylla;4~23,27~46:欧洲白桦与白桦的杂种F1代个体The F1 progeny from B. pendula and B. platyphylla crossing. |
利用FslinkageMao 1.0软件设置最大重组率为0.5和最小LOD值为3.0,通过对拟测交分离位点进行两点分析和多点连锁分析,分别构建了欧洲白桦和白桦的遗传连锁群。来自欧洲白桦的137个多态性位点,有105个位点分布于12个较大的遗传连锁群上(4个以上标记),连锁标记覆盖的总图距为1 533.3 cM,相邻标记间最大距离是46.8 cM,最小距离是1.3 cM,标记间平均距离为16.4 cM,还有12个位点分布在2个三连体和3个连锁对上(图 2a);来自白桦的136个多态性位点,有108个位点分布于12个较大的连锁群上(4个以上标记),覆盖的总图距为1 847.8 cM,相邻标记间最大距离是66.2 cM,最小距离是1.3 cM,标记间平均距离为19.2 cM,有2个三连体和4个连锁对(图 2b)。其余14个位点为非连锁位点,没有被定位在连锁群上。

|
图 2 欧洲白桦和白桦的RAPD标记连锁群 Figure 2 The linkage groups of B. pendula (a) and B. platyphylla (b) based on RAPD markers 连锁群的右侧为分子标记代号,左侧为遗传距离(cM)。 The codes on the right side of linkage group are the names composed by polymorphic DNA band; genetic distances in centiMorgans (cM) are given on the left side of linkage group. |
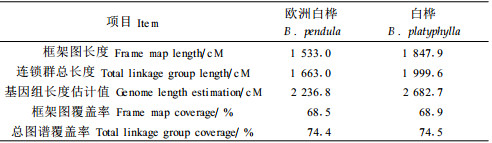
参照Chakravarti等(1991)的方法,估算欧洲白桦和白桦的连锁群总长度分别为1 663.0 cM和1 999.6 cM;估算欧洲白桦和白桦的基因组长度分别为2 236.8和2 682.7 cM,欧洲白桦框架图覆盖率和总图谱覆盖率分别为68.5%和74.4%,白桦框架图覆盖率和总图谱覆盖率分别为68.9%和74.5%(表 1)。
|
|
构建植物分子遗传连锁图谱是分子标记辅助选择育种基础。农作物的作图群体一般为近交F2、回交群体或DH群体等,这些群体的亲本连锁相已知,所有个体均可提供位点间的连锁信息,位点间连锁不平衡最大化。但是林木与此不同,大多数自交不亲合或近交衰退,遗传负荷一般较高,利用近交分离群体几乎不可能。但树木具有异交性和F1分离特性,可利用F1代作为树木的作图群体。作为用于研究林木遗传差异和构建林木遗传图谱的分子标记,由于RAPD和AFLP等随机标记具有简便快捷、灵敏度高、模板DNA需要量少及多态性丰富等优点,使之成为揭示林木遗传差异和构建林木遗传图谱的重要工具。据统计,一半以上的林木分子标记遗传连锁图谱是用RAPD和AFLP等随机标记构建而成的。但许多研究者发现RAPD和AFLP标记在林木杂种F1中普遍存在着偏分离和异常分离。本研究发现RAPD标记在欧洲白桦×白桦F1杂种中的测交位点是主体,占位点总数的93.2%,同时存在偏分离和异常分离现象。偏分离的原因目前有多种解释,主要是从试验误差、人为因素以及生物因素三方面着手。生物因素则包括染色体缺失、遗传分离机制、发育基因、隐性致死基因等(Bradshaw et al., 1994;Kuang et al., 1999)。本研究的作图群体为79株,作图群体小可能是造成偏分离现象发生的重要原因。偏分离位点可否用于图谱构建,在林木构图中没有明确的限定。早期的遗传图谱大多将偏分离位点用于遗传图谱构建(Bradshaw et al., 1994;Grattapaglia et al., 1994;苏晓华等,1998)。但偏分离位点往往会使连锁的检验受到影响,一些本来不连锁的标记位点由于偏分离位点的存在可能会出现连锁现象,一些本来连锁的位点由于偏分离的存在却无法检测到连锁。另外,偏分离标记位点的应用会导致连锁群上标记位点间距离不真实(Cervera et al., 2001)。本研究按拟测交策略,由于分析软件的限制,仅将符合1:1分离的位点用于连锁分析,而3:1分离的位点没有用于遗传连锁分析。试验中还发现双亲中无多态性的位点在子代中却发生了1:1分离。有研究认为,这类标记位点有可能成为2张亲本遗传连锁图谱连结的桥梁,识别并连结2个亲本的同源染色体,实现2张图的一体化(Maliepaard et al., 1998;Wu et al., 1999;张新叶等,2000)。但由于所发现的RAPD共显性标记位点较少,且没有合适的分析软件,因而没能将2张图谱进行整合。最近施季森等(2006)提出了利用林木F1代群体所有分离比的位点构建遗传图谱的新方法,充分考虑了不同分离比例位点以及位点间连锁相的信息,而且可以将亲本2张图谱整合到1张图谱上,使得构建高密度的林木遗传连锁图谱成为可能。总之,随着多种遗传标记方法的逐步完善和发展以及作图软件功能的增强,超高密度遗传连锁图谱构建和重要性状的精细定位以及分子标记辅助林木早期选择和基于遗传图谱的图位克隆将会很快在林木中实现,林木遗传图谱在林木育种改良研究中的重要地位更加突出,前景更为广阔。
姜静, 杨传平, 刘桂丰, 等. 2001a. 利用RAPD标记技术对白桦种源遗传变异的分析及种源区划. 植物研究, 21(1): 126-130. |
姜静, 杨传平, 刘桂丰, 等. 2001b. 利用RAPD标记技术对桦树种间亲缘关系的分析. 林业科学, 38(1): 154-156. |
李开隆, 姜静, 姜莹, 等. 2006. 白桦5×5完全双列杂交种苗性状的遗传效应分析. 北京林业大学学报, 28(4): 82-87. DOI:10.3321/j.issn:1000-1522.2006.04.016 |
施季森, 童春发. 2006. 林木遗传图谱的构建和QTL定位统计分析. 北京: 科学出版社.
|
苏晓华, 张绮纹, 郑先武, 等. 1998. 美洲黑杨×青杨分子连锁图谱的构建. 林业科学, 34(6): 29-37. DOI:10.3321/j.issn:1001-7488.1998.06.004 |
杨传平, 刘桂丰, 魏志刚, 等. 2004. 白桦强化条件下提早开花结实技术的研究. 林业科学, 40(6): 14-17. |
詹亚光, 李景云, 刘吉春, 等. 1994. 白桦嫩枝扦插繁殖技术的研究. 东北林业大学学报, 22(2): 6-10. |
詹亚光, 苏涛, 韩梅, 等. 2006. 转基因白桦外源基因的多重PCR快速检测. 植物研究, 26(4): 480-485. DOI:10.3969/j.issn.1673-5102.2006.04.022 |
张新叶, 尹佟明, 诸葛强. 2000. 利用RAPD标记构建美洲黑杨×欧美杨分子标记连锁图谱. 遗传, 22(4): 209-213. DOI:10.3321/j.issn:0253-9772.2000.04.004 |
朱翔, 刘桂丰, 杨传平, 等. 2001. 白桦种源区划及优良种源的初步选择. 东北林业大学学报, 29(5): 11-14. DOI:10.3969/j.issn.1000-5382.2001.05.003 |
Bradshaw H D Jr, Stettler R F. 1994. Molecular genetics of growth and development in Populus Ⅱ: Segregation distortion due to genetic load. Theor Appl Genet, 89: 551-558. |
Cervera M T, Storme V, Ivens B, et al. 2001. Dense genetic linkage maps of three Populus species (Populus deltoides, P. nigra and P. trichocarpa) based on AFLP and microsatellite markers. Genetics, 158: 787-809. |
Chakravarti A, Lasher L K, Reefer J E. 1991. A maximum likelihood for estimating genome length using genetic linkage data. Genetics, 128(1): 175-182. |
Grattapaglia D, Sederoff R. 1994. Genetic linkage maps of Eucalyptus grandis and Eucalyptus urophylla using a pseudotestcross: mapping strategy and RAPD markers. Genetics, 137: 1121-1137. |
Kuang H, Richardson T, Carson S, et al. 1999. Genetic analysis of inbreeding depression in plus tree of Pinus radiata D. Don. Ⅰ. Genetic map with distorted markers. Theoretical and Applied Genetics, 98: 697-703. DOI:10.1007/s001220051123 |
Maliepaard C, Alston F H, van Arkel G, et al. 1998. Aligning male and female linkagemaps of apple (Malus pumila Mill) using multialletic markers. Theor Appl Genet, 97: 60-73. DOI:10.1007/s001220050867 |
Pekkinen M, Varvio S, Kulju K K, et al. 2005. Linkage map of birth, Betula pendula Roth, based on microsatellites and ampliflied fragment length polymorphisms. Genome, 48(4): 619-625. DOI:10.1139/g05-031 |
Wu R L, Liu H X, Han Y F. 1999. Statistical methods for mapping quantitative trait loci in forest trees. Scient Silvae Sini, 35(2): 101-117. |