


2. 北京大学生命科学学院,北京100871
2. School of Life Sciences, Peking University, Beijing 100871, China
2014年诺贝尔化学奖授予发展超分辨率荧光显微成像技术的3位科学家,他们分别是美国霍华德·休斯医学研究所教授Eric Betzig、德国马克斯普朗克生物物理化学研究所教授Stefan W. Hell和美国斯坦福大学教授William E. Moerner。1 荧光显微镜的发明历程
光学显微镜自发明伊始,即与生命科学结下了不解之缘。16世纪末期,荷兰眼镜商Zaccharias Janssen和他的儿子把两个凸透镜放到一个镜筒中,结果发现镜筒能放大物体,这就是显微镜的前身。随后,荷兰人Anthony Von Leeuwenhoek通过精密研磨的玻璃透镜,首次将复式显微镜的分辨率提高到了300倍,并用它来观察牙缝中的微生物——细菌。后来,英国实验科学家Robert Hooke对这一显微镜进行了系统的改进,用它来观察软木塞的微小结构。他看到了上面致密排列的小室,并将其命名为细胞,记录在他1665年创作的《显微镜》一书中。然而,Robert Hooke与当时的科学巨匠牛顿产生了极大的分歧:牛顿认为光是粒子,而Robert Hooke则认为光是波。由于牛顿的学术地位和性格,使得当时的波动学说受到极大的排挤。
 | 图 1 2014年诺贝尔化学奖得主,从左至右依次为Eric Betzig,Stefan W. Hell和William E. Moerner(图片来源:Nobelprize.org) Fig. 1 The laureates of2014Nobel prize of Chemistry. Left to right: Eric Betzig,Stefan W. Hell,and William E. Moerner. |
1872年,德国数学和物理学家Ernst Abbe根据光的波动性,得出了以下结论:对一个理想的发光点,在经过显微镜光学系统后,将是一个高斯型的艾里斑。其光强分布的半高全宽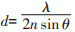 ,其中,λ是光的波长,n是折射率,θ是聚焦光锥的半角。
,其中,λ是光的波长,n是折射率,θ是聚焦光锥的半角。
 | 图 2 点扩展函数和阿贝分辨率极限 Fig. 2 Point spread function and Abbe criterion |
上述公式即阿贝公式,指明了3点:
1)由于光具有波动性,衍射将使得一个点不能被汇聚到另一个点(空间上没有大小的delta函数),而是具有一定的大小,这个大小就是我们常说的分辨率。
2)对于显微物镜,可以通过水、油等折射率比较高的液体,得到更好的分辨率。
3)由于sinθ最大值是1,在可见光下(波长400~700nm),能够获得的最小分辨率大约是200nm。
无论一个样品结构有多复杂,总是能够描述为是由一个个点构成的,因此,这个点的像描述了光学系统的响应函数,或者称之为系统的分辨率。该函数即点扩展函数。举例来说,在晴朗的夜空,人眼所看到的一颗星星的图像,就是人眼的点扩展函数。
通过阿贝公式,很容易想到的是,缩短波长即可产生更高的分辨率,比如利用X光。1986年,电子显微镜技术获得了诺贝尔物理学奖。在电子显微镜中,电子的德布罗意波长仅为光波的千分之一,属于X光,导致电子显微镜的分辨率达到0.1nm。这也进一步验证了阿贝的衍射理论在麦克斯韦方程下的普适性。1955年,美国细胞生物学家GeorgeEmil Palade利用电子显微镜发现了核糖体。核糖体是细胞内蛋白质翻译的工厂,它的发现对人类认识细胞功能迈出了重大的一步,George Emil Palade于1974年被授予诺贝尔生理学或医学奖。
然而,生物学家对于电镜可以说是爱恨交加。一方面,电镜使得过去不能够看到的样品细节能够被清晰地展现。但是,电镜却有着复杂、严苛的样品制备过程,如切片、脱水、固定、镀金等,这些均会破坏细胞的活性甚至改变其原有的结构。而且,电镜只能看到细胞内的结构,对于所观察到的结构的判断,则需要其他技术辅助辨别。
另一方面,光学显微需要所看的对象“色彩斑斓”,而细胞一般是高度透明的。为了能够看到细胞内部结构,人们发明了染色技术。最常用的染色技术——HE染色能够利用2种染料对生物组织进行染色,目前仍是临床采用的判定肿瘤良恶性的金标准。
然而,染色对于观察亚细胞结构仍存在较大挑战:染色是通过对光的直接吸收来产生的反差,也就是让细胞器有吸收,而周围没有吸收达到反差效果。光的吸收率可以用比尔定量描述:α=1-e-μl,其中μ为吸收系数,l是样品的厚度。当l非常小时,吸收率几乎为0,这也就是薄样品看上去几乎透明的原因。
传统的染色是吸收A波长,探测A波长的吸收率。为了解决这一问题,可以使系统吸收A波长,探测从样品染色上发出的B波长。这样,仅在有“染色”的地方发光,没有染色的地方是完全黑的,即荧光染色。1871年,Johann FriedrichWilhelm Adolf von Baeyer因合成荧光素获得1905年诺贝尔化学奖。在此基础上,能够特异性地标记细胞中的不同细胞器的荧光染色技术应运而生。2008年诺贝尔化学奖的绿色荧光蛋白能够通过外源性标记或者内源性转基因的方式选择性地让某一个感兴趣的细胞器发荧光,将活体成像往前推进了一大步。
对比荧光显微的图像和HE染色(图 3)可以发现,荧光染色在没有细胞器的地方是完全黑的,这就使得荧光显微有着传统染色无可比拟的高对比度。
 | 图 3 HE染色病理切片图(a)与细胞荧光显微图(b)的对比Fig. 3 Comparison of histological HE stain result (a) and cellular fluorescent imaging result (b) |
“工欲善其事,必先利其器”。要想探究生命的奥秘,首先就需要能够看清楚。然而不幸的是,很多亚细胞结构、细胞器和生物大分子的典型尺寸在几个到数百纳米,而光学显微的分辨率早在200年前就被阿贝公式限定在200nm,远远不能满足生命科学的需求。2 超分辨率荧光显微技术
传统的分辨可以定义为:如果点扩展函数较大,那么对于两个靠得很近的点,则不能分辨。在此基础上,实现超分辨的方法可分为两类:基于点扩展函数调制的超分辨技术,使得点扩展函数变小;基于随机单分子定位的超分辨技术,使得没有靠得很近的两个点同时发光。
基于点扩展函数调制的超分辨技术的代表为受激发射光淬灭(简称STED)技术。其基本思想是,在一个点扩展函数PSF1的基础上,用另一个环形点扩展函数PSF2去擦除PSF1的外围(抑制其外围发荧光),导致PSF1-PSF2剩下的光子为仅从PSF1中心发出,也就是点扩展函数变得更小。早在1994年,此次获奖的罗马尼亚裔德国科学家Stefan W. Hell(当时还是博士后)最先提出了打破光学衍射极限的构思,并最终于2000年在实验上得以实现[1]。在物理上,他通过受激辐射的原理,利用受激辐射进行环状擦除。由于这一方法所产生的点扩展函数不再受到衍射极限的限制,而仅仅取决于擦除的程度(擦除后剩下的区域大小),因此它的分辨率可表达为: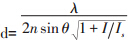 。从2000年开始,他不断改进STED技术,使其更加适用于生物研究。2006年,他展示了STED在绿色荧光蛋白上的应用[2];2008年,他实现了STED实时超分辨观察细胞囊泡的运动[3];2012年,他报道了利用STED进行活小鼠的神经突触生长过程的连续观测的成果[4]。另外,还通过其他光调制原理发明了一系列的超高分辨率技术,统称为可逆饱和荧光跃迁(RESOLFT),为超分辨率荧光显微成像技术的发展做出了巨大贡献。
。从2000年开始,他不断改进STED技术,使其更加适用于生物研究。2006年,他展示了STED在绿色荧光蛋白上的应用[2];2008年,他实现了STED实时超分辨观察细胞囊泡的运动[3];2012年,他报道了利用STED进行活小鼠的神经突触生长过程的连续观测的成果[4]。另外,还通过其他光调制原理发明了一系列的超高分辨率技术,统称为可逆饱和荧光跃迁(RESOLFT),为超分辨率荧光显微成像技术的发展做出了巨大贡献。
 | 图 4 STED超分辨率成像原理示意图 Fig. 4 Principle of STED super-resolution microscopy |
基于结构照明原理的超高分辨率技术是美国科学家Mats Gustafsson于2000年发明的,非常适于细胞研究,可惜分辨率只能提高1倍。该技术是基于两个高空间频率的图案重叠可以形成低频率莫尔条纹的原理,通过解析低频莫尔条纹实现超高分辨率成像。在此基础上,Mats Gustafsson发明了饱和SIM,能够利用荧光饱和实现更高的分辨率,其本质也是点扩展函数的缩小。可惜Mats Gustafsson于2011年51岁时因癌症去世,无缘分享此次诺贝尔奖。
基于随机单分子定位的超分辨技术的核心是,如果图像上的点不是同时亮起来,也就是不会有两个靠得很近的点同时亮,就可以通过定位的方式实现超分辨。虽然一次定位只能得到少数几个分子,但是通过数千张图片对数十万个单分子的定位,就可以获得一张高分辨率的图像。
 | 图 5 基于随机单分子定位的PALM/STORM超分辨原理图 Fig. 5 Diagram of the localization based PALM/STORM super-resolution microscopy |
这一超分辨技术发明于2006年,由本次诺贝尔奖得主Eric Betzig(光活化定位显微术PALM技术)[5]、哈佛大学教授庄小威(随机光学重构显微术STORM技术)[6]、以及SamuelHess(荧光活化定位显微术fPALM技术)[7]3个研究组分别同时独立发明,分别发表于Science,Nature Methods和Biophysical Journal。3种技术的原理非常像,都是基于荧光分子的光转化能力和单分子定位,通过用光控制每次仅有少量随机离散的单个荧光分子发光,并准确定位单个荧光分子点扩展函数的中心,通过多张图片叠加形成一幅超高分辨率图像。
William E. Moerner是单分子荧光技术的先驱人物。他在1989年任职于美国IBM研究中心时在世界上首次实现了单个分子的光吸收的测量[8],并在1997年与发展绿色荧光蛋白技术而获得2008年诺贝尔化学奖的钱永健合作,发现了绿色荧光蛋白的光转化效应[9]。而Eric Betzig是荧光显微技术领域的领军人物。他在1994年提出了基于单分子信号实现超高分辨率成像的思想[10],并于2006年在实验中得以实现。庄小威作为STORM超分辨技术的发明人,一直领导并推进着超高分辨率显微技术的发展和应用,是近8年来这个领域最活跃的研究团队。3 我国超分辨率光学显微技术发展现状
在中国,目前有数十家单位在进行超分辨率显微方面的研究工作,在研究上紧跟国际潮流。比如,本文作者北京大学席鹏于2010年首次在国内实现了STED超分辨[11,12,13],其后浙江大学刘旭[14]、中国科学院化学研究所方晓红[15]等课题组也报道了STED超分辨技术的进展。在PALM/STORM研究方面,北京大学孙育杰课题组一直在进行单分子成像和层状光成像方面的研究[16, 17],中国科学院生物物理研究所徐涛、徐平勇课题组研发新型光开关蛋白[18]、华中科技大学黄振立课题组开发新型定位算法等[19]。在结构光成像方面,中国科学院西安光学精密机械研究所姚保利课题组报道了基于DMD光调制的SIM[20]。在高速并行三维超分辨成像方面,席鹏课题组最近报道了一系列基于SOFI的超分辨技术[21, 22]。
当前,我国在超分辨率显微技术方面,已经有一支完善的研究队伍。摆在我们面前的首要问题是如何结合这些超分辨技术和其他光学技术,取长补短,实现更清(更高空间分辨率)、更快(更高时间分辨率)、更深(组织三维成像)方面的突破,向着活体三维动态超分辨方向不断推进。4 未来:更清、更快、更深
超分辨率显微作为一类很新的技术,突破了光学成像中的衍射极限,把传统成像分辨率提高了10~20倍,成为研究细胞结构的利器。过去七八年间,这些技术不断推进,先后实现了多色、三维和活细胞高速成像。其生物应用也很广泛,包括细胞膜蛋白分布、细胞骨架、线粒体、染色质和神经元突触等。超高分辨率技术一经出现就引起广泛关注,先在2006年被Science评为年度10大技术突破,随后被生物医学方法学最好的期刊Nature Methods评为2008年度方法。在2014年9月Nature Methods的10周年特刊评出的10年10大技术中,超高分辨率成像和单分子技术也都出现在榜中。
科技的进步,有赖于站在巨人的肩膀上,将其他技术有机地与自己的技术相结合。2005年的《生物技术当今观点》刊文指出了过去的光学显微的方法,如宽场、共聚焦、多光子、非线性、全内反和今天的超分辨技术之间的演进关系[23]。虽然经过10年的发展,超分辨显微在三维空间分辨率的提升上有了长足进步,然而这往往是以牺牲时间分辨率为代价的。未来的技术进步,需要在并行化、高速、活细胞和活组织方面,实现超高时空分辨活体成像。最近,Hell课题组将所有的超分辨技术——STED的环形光、SIM的条纹、PALM/STORM的光开关蛋白有机结合,实现了并行的超分辨,将超分辨率显微的速度大幅提升[24, 25]。
历史总是在自我重复中不断前进。在1903年,RichardZsigmondy发明了超显微镜,这一显微技术因其分辨率能够突破波长尺度而获得1925年诺贝尔奖。由于它提供了全新的研究问题的尺度,人们甚至将它看作是纳米科技的起始[26]。在2004年,这一技术在层状光照明显微中得到新的应用,同时被评为Nature Methods10大年度方法[27]。那么,古老的诺奖超显微技术,会不会和今天的诺奖超分辨率荧光显微擦出新的思想火花呢?
后记:写下初稿的时候刚好是诺贝尔奖公布的时间。2014年11月,Eric Betzig发表在Science的工作,将Light sheet与超分辨结合,对活体胚胎进行成像[28]。这一技术向更清、更快、更深迈出了新的一步,也印证了我们文终提出的问题。
| [1] | Hell S W, Wichmann J. Breaking the diffraction resolution limit by stimulated emission: stimulated-emission-depletion fluorescence microscopy[J]. Optics Letters, 1994, 19(11): 780-782. |
| [2] | Willig K I, Kellner R R, Medda R, et al. Nanoscale resolution in GFPbased microscopy[J]. Nature Methods, 2006, 3(9): 721-723. |
| [3] | Westphal V, Rizzoli S, Lauterbach M, et al. Video-rate far-field optical nanoscopy dissects synaptic vesicle movement[J]. Science, 2008, 320 (5873): 246. |
| [4] | Berning S, Willig K I, Steffens H, et al. Nanoscopy in a living mouse brain [J]. Science, 2012, 335(6068): 551-551. |
| [5] | Betzig E, Patterson G H, Sougrat R, et al. Imaging Intracellular Fluorescent Proteins at Nanometer Resolution[J]. Science, 2006, 313(5793): 1642-1645. |
| [6] | Rust M J, Bates M, Zhuang X. Sub-diffraction-limit imaging by stochastic optical reconstruction microscopy (STORM)[J]. Nature Methods, 2006, 3 (10): 793-796. |
| [7] | Hess S T, Girirajan T P K, Mason M D. Ultra-High Resolution Imaging by Fluorescence Photoactivation Localization Microscopy[J]. Biophysical Journal, 2006, 91(11): 4258-4272. |
| [8] | Moerner W, Kador L. Optical detection and spectroscopy of single molecules in a solid[J]. Physical Review Letters, 1989, 62(21): 2535. |
| [9] | Dickson R M, Cubitt A B, Tsien R Y, et al. On/off blinking and switching behaviour of single molecules of green fluorescent protein[J]. Nature, 1997, 388(6640): 355-358. |
| [10] | Betzig E. Proposed method for molecular optical imaging[J]. Optics Letters,1995,20(3):237-239. |
| [11] | Liu Y, Ding Y, Alonas E, et al.Santangelo P J, Jin D, et al. Achieving λ/10 Resolution CW STED Nanoscopy with a Ti: Sapphire Oscillator[J]. PLoS One, 2012, 7(6): e40003. |
| [12] | Xie H, Liu Y, Santangelo P J, et al. Analytical description of highaperture STED resolution with 0-2pi vortex phase modulation[J]. Journal of Optical Society of America A, 2013, 30(8): 1640-1645. |
| [13] | Yang X, Tzeng Y-K, Zhu Z, et al. Sub-diffraction imaging of nitrogenvacancy centers in diamond by stimulated emission depletion and structured illumination [J]. RSC Advances, 2014, 4: 11305-11310. |
| [14] | Wang Y, Kuang C, Li S, et al. A 3D aligning method for stimulated emission depletion microscopy using fluorescence lifetime distribution[J]. Microscopy Research and Technique, 2014, 77(11): 935-940. |
| [15] | Yu J, Yuan J, Zhang X, et al. Nanoscale imaging with an integrated system combining stimulated emission depletion microscope and atomic force microscope[J]. Chinese Science Bulletin, 2013, 58(33): 4045-4050. |
| [16] | Zong W, Zhao J, Chen X, et al. Large-field high-resolution twophoton digital scanned light-sheet microscopy[J]. Cell Research, 2014, 25: 254-257. |
| [17] | Liu Z, Xing D, Su Q P, et al. Super-resolution imaging and tracking of protein-protein interactions in sub-diffraction cellular space[J]. Nature Communications, 2014, 5: 4443. |
| [18] | Chang H, Zhang M, Ji W, et al. A unique series of reversibly switchable fluorescent proteins with beneficial properties for various applications[J]. Proceedings of the National Academy of Sciences, 2012, 109(12): 4455-4460. |
| [19] | Quan T, Li P, Long F, et al. Ultra-fast, high-precision image analysis for localization-based super resolution microscopy[J]. Optics Express, 2010, 18(11): 11867-11876. |
| [20] | Dan D, Lei M, Yao B, et al. DMD-based LED-illumination Superresolution and optical sectioning microscopy[J]. Scientific Reports, 2013, 3: 1116. |
| [21] | Chen X, Zeng Z, Wang H, et al. Three dimensional multimodal sub-diffraction imaging with spinning-disk confocal microscopy using blinking/fluctuation probes[J]. Nano Research, 2015: doi: 10.1007/ s12274-015-0736-8. |
| [22] | Zeng Z, Chen X, Wang H, et al. Fast Super-Resolution Imaging with Ultra-High Labeling Density Achieved by Joint Tagging Super-Resolution Optical Fluctuation Imaging[J]. Scientific Reports, 2015, 5: 8359. |
| [23] | Garini Y, Vermolen B J, Young I T. From micro to nano: recent advances in high-resolution microscopy[J]. Current Opinion in Biotechnology, 2005, 16(1): 3-12. |
| [24] | Chmyrov A, Keller J, Grotjohann T, et al. Nanoscopy with more than 100,000 'doughnuts'[J]. Nature Methods, 2013, 10(8): 737-740. |
| [25] | Chen X, Xi P. Hundred-thousand light holes push nanoscopy to go parallel[J]. Microscopy Research and Technique, 2014, 78(1): 8-10. |
| [26] | Mappes T, Jahr N, Csaki A, et al. The Invention of Immersion Ultramicroscopy in 1912—The Birth of Nanotechnology[J] Angewandte Chemie International Edition, 2012, 51(45): 11208-11212. |
| [27] | Huisken J, Swoger J, Del Bene F, et al. Optical sectioning deep inside live embryos by selective plane illumination microscopy[J]. Science, 2004, 305(5686): 1007-1009. |
| [28] | Chen B C, Legant W R, Wang K, et al. Lattice light-sheet microscopy: Imaging molecules to embryos at high spatiotemporal resolution[J]. Science, 2014, 346(6208): 1257998. |