 2021, Vol. 41
2021, Vol. 41



2. 南京大学环境学院, 南京 210023
2. School of Environment, Nanjing University, Nanjing 210023
偶氮污染物(Azo Contaminants, ACs)是以氮氮双键为核心结构的有机分子, 是常见的化学试剂、化工中间体和终端产品, 在pH检测、非线性光学材料合成(Samanta et al., 2018)、活性氧自由基(Reactive Oxygen Species, ROS)鉴定及日用品染色等方面有广泛应用.偶氮污染物对生态系统具有不良影响, 其次生产物—还原性苯胺类化合物更是具有“三致”作用.因此, 有关偶氮污染物的处理处置成为较受关注的研究方向(Petrella et al., 2013).常见的方法有高级氧化、光催化、吸附、生态修复等(Yang et al., 2014; Zhao et al., 2019).目前, 人们对偶氮污染物的光降解研究通常着重于讨论偶氮化合物与功能材料的相互作用(Petrella et al., 2013; Yang et al., 2014; Cai et al., 2017; Guan et al., 2019; Zhao et al., 2019), 针对其本身与光子的相互作用鲜被关注, 然而这既是光反应的核心机理之一, 也是污染物浓度定性定量分析的理论基础.因此, 研究“光子-偶氮污染物结构-电子激发”间的耦合模式可以填补空白, 为环境监测、环境修复、环境功能材料开发及非线性光学材料合成提供理论依据.Gagliardi等(2004)通过理论计算发现了偶氮苯分子光致异构过程中低能单线态的存在, 从而解释了偶氮苯光化学的波长依赖现象, 为光催化波长、光源及介质的选用提供了依据.Döbbelin等(2016)通过对偶氮苯分子结构的调控, 在365 nm光照下成功剥离石墨晶体得到了单层石墨烯, 在环境友好的非强酸强碱条件下实现了石墨烯制备.此类研究为光照环境中污染物迁移转化、碳基环境材料的开发建立了良好的理论基础.
偶氮污染物分子与光子相互作用的具象化表现为紫外-可见吸收光谱(下称紫外光谱), 在一定浓度范围内, 吸光度与浓度具有良好的线性关系.实验紫外光谱具有较长的研究历史, 是定性定量、监控反应过程的有力工具.溶剂环境中, 一般分辨率的紫外光谱通常被认为缺乏精细解析能力, 溶剂拉平效应、氢键或“溶质-溶剂”络合物的形成模糊了精细结构.已有研究使用多高斯峰拟合(Zhang et al., 2012)、差分光谱(Yan et al., 2016)等技术对分子构象或复杂结构中官能团分布进行探索, 但离精细结构解析仍有差距.计算化学可以通过振子强度与激发能研究吸收峰强度与位置, 是有力的光谱指认工具.通常认为偶氮分子中n电子跃迁至π反键轨道(n→π*)是最容易发生的跃迁过程(激发能大约为3.00 eV), 但此过程激发电子(n电子)来源于2个氮原子, 其占据整个分子的摩尔比例为8.33%(以偶氮苯为例), 此局域激发过程与实验光谱主要吸收峰建立关联需要更明确的实验或理论计算证据.Daniel等(2011)在PBE0/TZVP计算水平下研究了6种偶氮染料的紫外吸收光谱, 指出n→π*跃迁尽管是最容易发生的跃迁, 但其吸收(振子)强度为0(跃迁禁阻), 因此不对实验光谱第一显著吸收峰做贡献, 很多理论研究都持有类似观点(Manzoni et al., 2019).但若因此完全否认偶氮键对紫外吸收的贡献, 亦缺乏明确的科学证据.
基于此, 本研究以偶氮苯和甲基橙为目标污染物, 通过对比“顺反式”偶氮苯分子的实验与计算光谱, 研究不同共轭状态下偶氮键的紫外吸收特性, 阐明“偶氮键”对“偶氮污染物”吸收光谱的贡献情况, 研究弱相互作用对吸收光谱的影响.从分子轨道、振子强度等角度, 探明吸收光谱与电子分布之间的关系, 揭示显色行为内在机理, 为偶氮污染物紫外光谱指认、污染物光催化、污染物环境行为及分子开关设计等研究提供理论依据.
2 材料与方法(Material and methods) 2.1 实验方法研究使用的两种偶氮污染物分别为偶氮苯(CAS No.103-33-3, 上海阿拉丁)和甲基橙(CAS No.547-58-0, 上海阿拉丁), 除乙醇(上海国药)为色谱纯外, 其他试剂均为分析纯(上海国药).实验紫外光谱测量波长为200~600 nm(上海元析UV8000-S, 10 mm石英比色皿), 使用0.1 mol·L-1 HCl和NaOH调节被测溶液pH到2、7和12, 污染物浓度为10 μmol·L-1, 溶剂为1%乙醇水溶液(助溶), 实验用水电阻率为18.2 MΩ·cm-1, 所有实验均在(10±2) ℃下进行.为了便于实验室间对比, 后续补充了(22±2) ℃下的实验光谱, 与10 ℃下的实验光谱相比, 其峰偏移小于0.1%, 不存在显著区别.
2.2 理论计算方法本文的计算工作主要包括以下部分: ①基态分子构型的优化和振动分析, 以获得稳定结构;②以①为基础的理论紫外光谱指认(使用含时密度泛函方法);③分子动力学模拟, 建立团簇模型, 预测紫外吸收光谱.其中, ①和②使用高斯09w计算(Frisch et al., 2009), ③的分子动力学部分使用Gromacs2018.8模拟(Abraham et al., 2015).波函数分析使用Multiwfn进行(Lu et al., 2012).除非特别声明, ①和②均在SMD模型的水溶液环境中进行计算并使用GD3(BJ)方法进行色散校正(Marenich et al., 2009; Grimme et al., 2011), 以满足弱相互作用的计算需求.鉴于M06-2x和PBE38分别在结构优化和大共轭分子激发态研究中的良好表现(Goerigk et al., 2010; Walker et al., 2013), ①中使用M06-2x/def2-tzvp进行优化和振动分析以获得局部最稳定的分子构型, ②中使用PBE38/def2-tzvpp进行含时密度泛函(Time-Dependent Density Functional Theory, TDDFT)计算以获得理论紫外吸收光谱, 为尽可能覆盖感兴趣的光谱范围, 计算激发态数设置为100.
分子动力学模拟在10 nm边长的周期性立方体水盒子中进行, 水分子使用SPC/E模型, 水分子和污染物分子模拟参数分别由琥珀14SB力场(Amber14SB)和通用琥珀力场(General amber force field, GAFF)定义(Maier et al., 2015, Wang et al., 2004), 原子电荷为限制性静电势拟合电荷(0.5)(Restrained Electrostatic Potential, RESP2(0.5))(Schauperl et al., 2020).初始结构使用Packmol 18.169搭建(Martinez et al., 2009).
3 结果与讨论(Results and discussion) 3.1 不同共轭环境中偶氮键电子跃迁特性偶氮苯(Azobenzene, AB)是偶氮染料分子的核心结构, 具有顺式与反式两种构型(图 1A、1B), 含有不同共轭状态下的偶氮键(顺式→局部共轭, 反式→全局共轭).顺式能量高于反式, 在特定波长光线“激发”下, 反式可以转化为顺式, 撤去激发源后, 顺式通过热弛豫回到反式, 因此, 反式是环境中的主要构型(Gagliardi et al., 2004; Duarte et al., 2014).位阻效应、色散力及异构化能垒(Cattaneo et al., 1999; Duarte et al., 2014; Titov et al., 2015)共同导致顺式偶氮苯能在较高能级相对稳定存在.顺式和反式具有类似的轨道分布特征, 即两者的最高占据轨道(Highest Occupied Molecular Orbital, HOMO)和最低未占据轨道(Lowest Unoccupied Molecular Orbital, LUMO)都对应各自的48、49号分子轨道.前线分子轨道体现了反式偶氮苯的整体芳香性(图 1a~1d), 其HOMO轨道体现了苯环与偶氮键结构中沿分子平面对称分布的电子结构(图 1c), 其HOMO-1轨道体现出偶氮键附近σ键和孤对电子(图 1d), 其反键轨道亦具有显著的芳香特征(图 1a、1b).
 |
| 图 1 顺反式偶氮苯结构与前线分子轨道等值面图 (0.05 a.u.)(白(蓝)椭球体分别代表正(负)相位, 括号中数字为分子轨道序号) Fig. 1 Cis-trans azobenzene structure and Iso-value surface diagram of its frontier molecular orbital (0.05 a.u.)(white (blue) ellipsoid represents positive (negative) phase, respectively. Numbers in brackets are the molecular orbital number) |
顺式偶氮苯HOMO轨道中可以观察到多种电子结构的混合现象(图 1g), 苯环上具有较强的芳香性特征, 在偶氮键上能够观察到n电子结构(图 1g中偶氮键上方两个等值面椭球体), 而在连接苯环与偶氮键的C—N键上则观察到π-σ的混合结构特征(图 1g中苯环平面两侧的等值面椭球体和毗邻C—N键上沿轴向分布的等值面椭球体).整体而言, 顺式结构的波函数构造显得更加“凌乱”和“破碎”.由此可见, 顺式偶氮苯的整体芳香性弱于反式偶氮苯, 偶氮键中的n电子(或π电子)在顺式结构中可能不参与全局共轭.换言之, 偶氮键在反式偶氮苯中处于一个“大”共轭体系中, 顺式则不是.为了进一步验证偶氮键的共轭环境, 针对性地考察π电子分布情况, 引入局部轨道定位函数(Localized Orbital Locator, LOLπ)(Schmider et al., 2000; Lu et al., 2020), LOL是基于电子动能密度(Kinetic Energy Density, KED)的概念密度泛函函数, 能清晰直观地展现体系中的化学键区域(图 2b、2d).在两种结构中, π电子密度分布特征类似, 均可粗略划分为两部分: 苯环π电子及偶氮键π电子(图 2a、2c), 但无法判断它们之间的沟通(共轭)情况.LOLπ函数等值面表明, 反式偶氮苯中所有π电子形成了一个完整的“大”共轭结构(图 2b), 体现出偶氮键桥接效应.顺式偶氮苯中的π电子则形成3个相对独立的部分(图 2d), 因此, 两者激发特性必有差异.
 |
| 图 2 偶氮苯π电子密度等值面图(0.05 a.u.)(a, c)与LOLpi函数等值面图(0.35 a.u.) (b, d) Fig. 2 Isosurface of azobenzene π electron density iso-value surface(0.05 a.u.)(a, c) and LOLpi function iso-value surface (0.35 a.u.)(b, d) |
鉴于PBE38(HF成分37.5%)理论方法在共轭体系激发态研究中的良好表现(Goerigk et al., 2010; Liu et al., 2020a; 2020b), 本研究使用此理论方法对两种偶氮苯分子进行TDDFT计算, 得到的理论计算光谱与实验光谱高度重合, 为后续的指认工作奠定了良好基础(图 3a).全局芳香性使得π电子能在整个分子中相对自由地移动, 促使激发能降低、吸收峰红移.引入反式偶氮烯分子( 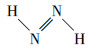
 |
| 图 3 偶氮苯实验与计算紫外-可见吸收光谱 (a.不同pH条件下反式偶氮苯分子实验和计算光谱, b.顺(反)偶氮苯计算光谱, exp.为实验光谱, theo.为计算光谱) Fig. 3 Experimental and theoretical UV-Vis spectrum of azobenzene (a.Experimental and theoretical spectrum of trans-azobenzene under different pH conditions, b.Theoretical spectrum of cis (trans) azobenzene) |
| 表 1 偶氮烯和顺反偶氮苯激发态相关指数(计算得到) Table 1 Corresponding index of the excited states of diazene and cis-trans azobenzene (calculated) |
水环境中反式偶氮苯与顺式偶氮苯的E1分别为2.775 eV(447 nm)和2.897 eV (428 nm), 证明π电子的全局共轭有利于S0→S1激发能的降低, 而较小的振子强度表明反式偶氮苯实验光谱中320 nm处的最大吸收峰(图 3a)由其他跃迁过程贡献.反式偶氮苯的计算光谱中未能观察到相应的447 nm的强吸收峰(图 3b黑线), 然而其实验光谱在422 nm位置观察到一个弱的展宽较大的吸收峰(图 3a中422 nm绿框附近), 此处吸收存在多种可能性: 有研究认为此处吸收是偶氮苯质子化导致(童沈阳, 1980), 也有可能是跃迁磁偶极矩贡献(0.974 a.u.), 同时不排除顺式偶氮苯的存在, 偶氮苯顺反式异构化能约为1.09 eV (25.136 kcal·mol-1)(Yu et al., 2015), 系统中有可能存在少量顺式偶氮苯, 实验光谱较深紫外区的吸收峰亦证明了这一点(图 3a中红框229 nm处吸收峰), 与顺式偶氮苯计算光谱高度一致(图 3b中230 nm处), pH=12时229 nm处高斯峰形破缺由高浓度Na+导致, 因为pH=12对应浓度的Na+(10 mmol·L-1 NaOH)在 < 210 nm波长时存在显著紫外吸收, 与污染物的峰叠加后导致原本对称的峰形失去了对称性.
分子激发过程是电子转移过程, 将激发前电子位置定义为“空穴”, 将激发后电子位置定义为“电子”(Liu et al., 2020c), 就可以直观地考察电子转移.环境化学中关心波长范围一般为200~600 nm, 对应的顺式偶氮苯跃迁过程包括S0→S1到S0→S10.在这10个跃迁过程中, 最低对应S0→S1(428 nm, f=0.031), 振子强度大于0.1的跃迁过程包括S0→S2(247 nm)与S0→S6(231 nm)(表 1), 是最需要关注的3个跃迁.与之对应的, 反式偶氮苯应关注的激发态为S1、S2及S6(表 1).图 4中清楚地展现了顺式偶氮苯3个激发过程中的电子变化.参与S0→S1跃迁的电子在被“激发”之前主要分布在偶氮键沿C—N键位置, 由N原子及C—N键成键电子组成(图 4a), “激发”后的电子呈现出非常典型的π分布特征, 应将其指认为n→π*激发.对于“电子”而言, 此计算级别下LUMO轨道的贡献率超过了94%;对于“空穴”而言, 主要贡献轨道为HOMO轨道(48号分子轨道), 贡献率为78%.因此, 顺式偶氮苯的S0→S1跃迁主要对应其HOMO-LUMO轨道跃迁;S0→S2和S0→S6是能量更高的激发过程, 从空穴-电子等值面考察基本可以指认为π→π*激发(图 4b、4c), 但仍然包括部分n→π*成分.综上所述, 在非全局共轭体系中, 偶氮键第一激发态为n→π*, 但振子强度较弱(f=0.031), 表明“孤立”偶氮键对顺式结构中的紫外吸收贡献有限, 即实验光谱中测得的第一个主要吸收峰不是由S0→S1贡献, 不能指认为n→π*跃迁类型.
 |
| 图 4 顺式偶氮苯S0→S1、S0→S2、S0→S6激发时电子分布变化 (MO是分子轨道Molecular Orbital的缩写, MO之后的数字代表轨道序号;a、b、c分别对应第1、2、6激发态;空穴和电子等值面图 (0.002 a.u.)中白色和红色椭球体分别代表“电子”和“空穴”;分子轨道等值面图(0.05 a.u.)中白色和红色椭球体分别对应正负相位) Fig. 4 The electron distribution of cis azobenzene during the excitation of S0→S1, S0→S2, S0→S6 (MO is the abbreviation of Molecular Orbital, and the number after MO represents the orbital number. In the "electron-hole" column, the white and red ellipsoids represent the "holes" and the "electron" isosurface (0.002a.u.) respectively. In the "MO" column, the white and red ellipsoids represent the positive and negative phases of the wave function (0.05 a.u.), respectively) |
反式偶氮苯是环境中的主要构型, 研究其实验和计算光谱对偶氮类污染物检测和光催化波长选用具有指导意义.由图 3a可知, 计算光谱很好地吻合了实验光谱, 其波长误差仅为6 nm.反式偶氮苯的两个苯环具有很强的共面性(二面角179.99°), 之前也证明了其整体芳香性(图 1b、1c和图 2a、2b). 推测偶氮苯分子的第一个“显著”吸收峰(图 3a实验光谱320 nm处峰)必然有π电子激发参与.“空穴-电子”分析表明, 反式偶氮苯的S0→S1激发主要由偶氮键参与, 具有显著的n电子和σ电子特征, 激发能为2.775 eV, 中心峰位置在447 nm处, 且为跃迁禁阻(f=0), 其“空穴”和“电子”具有显著的π对称性(图 5a).反式偶氮苯的S0→S2跃迁是事实上的HOMO→LUMO跃迁, 分别对应第48号到49号分子轨道跃迁(顺式的S0→S1跃迁对应HOMO→LUMO跃迁), 其激发能为3.917 eV, 中心峰位于316.57 nm处, 与实测峰位置(320 nm)偏差小于5%.反式偶氮苯的S0→S2激发有很多“大共轭”结构的π电子参与, 激发轨道组成相对单纯, “空穴”和“电子”主要由48号和49号轨道贡献(图 5b).48号轨道波函数等值面图表明苯环和偶氮键上的π电子均对S0→S2激发做出了重要贡献, 且激发后电子仍呈较强的π对称性(图 5b).由此可见, 反式偶氮苯的实验第一吸收峰应当指认为π→π*跃迁, 且主要官能团—苯环和偶氮键都有贡献, 而非只有后者贡献;与之相反, 偶氮键激发过程具有很强的局域特征, 吸收强度很弱, 实验光谱未能体现.由此可见, 偶氮键在电子激发过程中发挥了“桥接作用”, 能够促进反式偶氮苯吸收峰红移.
 |
| 图 5 反式偶氮苯S0→S1、S0→S2、S0→S6激发时电子分布变化 (MO是分子轨道molecular orbital的缩写, MO之后的数字代表轨道序号;a、b、c分别对应第1、2、6激发态;空穴和电子等值面图 (0.002 a.u.)中白色和绿色椭球体分别代表“电子”和“空穴”;分子轨道等值面图(0.05 a.u.)中白色和绿色椭球体分别对应正负相位) Fig. 5 The electron distribution of trans-azobenzene during the excitation of S0→S1, S0→S2, S0→S6 (MO is the abbreviation of Molecular Orbital, and the number after MO represents the orbital number. In the "electron-hole" column, the white and green ellipsoids represent the "holes" and the "electron" isosurface (0.002a.u.) respectively. In the "MO" column, the white and green ellipsoids represent the positive and negative phases of the wave function (0.05 a.u.), respectively) |
甲基橙阴离子的紫外光谱受到多种因素影响, 其中, 溶剂化、络合态及与离子的相互作用是重要的影响因素.为了尽可能体现甲基橙在水溶液中的真实行为, 本研究对多种结构进行了考察, 通过与实验光谱的对比, 回归得到“真实”结构.分子表面静电势(图 6)表明, 甲基橙阴离子(Methyl Orange Anion, MOA)分子的长轴方向存在明显的电势差异, 处于显著极化状态, 这是因为“推拉电子”基团存在, 导致核心“偶氮苯”结构高度极化, 且磺酸基具有很强的亲水性, 所以MOA分子可能与周围溶剂环境因子存在“复杂”的弱相互作用, 导致其吸收峰红移.作为MOA的抗衡离子, 钠离子易与MOA表面低电位点发生静电作用, 除磺酸基附近的低电势区域, MOA在其偶氮键附近亦存在较多的极小值点, 平均静电势约为-75 kcal·mol-1, 亦有可能和钠离子相互吸引.在10 nm边长的水盒子中, 加入20个MOA分子进行动力学模拟(图 7a), 结果表明在10 ns后就能观察到二聚体和多聚体的形成(图 7b、7c).作为团聚的起点, 二聚结构之间的相互作用能揭示MOA分子聚合的原始驱动力, 也能用于研究分子间相互作用对光谱的影响.基于静电势分析和分子动力学模拟结果(图 7), 选择以下8种MOA体系进行TDDFT研究: 甲基橙阴离子单体(MOA)、二聚体(MOA Dimer, MOAD, 来自分子动力学模拟结果)、含一个钠离子的MOA体系(Sodium cation-MOA, SCMOA)、含两个钠离子的MOA体系(SC2MOA)及含一个钠离子及显式水系统的MOA体系(SCMOA_WaterCluster, SCMOA_WC)(图 8).
 |
| 图 6 甲基橙分子表面静电势 (阴离子, 水溶液环境)(红色离散小球为局部静电势极小值点, 甲基橙分子中灰色、蓝色、黄色、红色、白色圆球体分别对应碳、氮、硫、氧、氢原子) Fig. 6 Electrostatic potential of methyl orange (anion, aqueous environment)(The red discrete spheres are the Localized ESP minima point (LEMP). The gray, blue, yellow, red, and white spheres within MOA molecular correspond to atoms of carbon, nitrogen, sulfur, oxygen, and hydrogen) |
 |
| 图 7 甲基橙阴离子分子动力学模拟结果 (a.分子动力学模拟时间线上的帧结构, b.10 ns时模拟得到的二聚体结构;c.50 ns时模拟得到的五聚体结构;灰色、蓝色、黄色、红色、白色圆球体分别对应碳、氮、硫、氧、氢原子) Fig. 7 Molecular dynamics simulation results of methyl orange anion (a.Frame structure in the timeline of molecular dynamics simulation, b, The dimer structure obtained at 10 ns, c.The pentamers structure obtained at 50 ns. The gray, blue, yellow, red, and white spheres within MOA molecular correspond to atoms of carbon, nitrogen, sulfur, oxygen, and hydrogen) |
 |
| 图 8 8种用于紫外光谱研究的甲基橙模型 (灰色、蓝色、黄色、红色、白色、褐色圆球体分别对应碳、氮、硫、氧、氢、钠(虚线圈出的)原子;各体系命名法如下: 甲基橙阴离子单体(MOA)、二聚体(MOA Dimer, MOAD)、含一个钠离子的MOA体系(Sodium cation-MOA, SCMOA)、含两个钠离子的MOA体系(SC2MOA)及含一个钠离子及显式水系统的MOA体系(SCMOA_WaterCluster, SCMOA_WC)) Fig. 8 Eight methyl orange models for UV-Vis absorption spectrum study (The gray, blue, yellow, red, white, and brown spheres correspond to atoms of carbon, nitrogen, sulfur, oxygen, hydrogen, and sodium (circled by dotted lines). The nomenclature of each system is as follows: Methyl orange anion monomer (MOA), Dimer (MOA Dimer, MOAD), MOA system containing one Sodium ion (Sodium cation-MOA, SCMOA), MOA system containing two sodium ions (SC2MOA) and MOA system containing one sodium ion and an explicit water system (SCMOA_WaterCluster, SCMOA_WC)) |
当pH=7或12时, 甲基橙主要以阴离子形态赋存, 此时的实验光谱主要由MOA贡献.实验光谱第一个显著吸收峰出现在大约463 nm处, 其次在278 nm处(图 9a).除深紫外区(< 250 nm), pH=12和pH=7的实验光谱几乎完全重合.从MOA的实际形态考虑(-1价), 计算MOA光谱时应该使用弥散函数, 但会导致计算量激增(超过了本研究可调用的计算资源)和自洽场收敛困难, 就本研究中TDDFT计算而言, 现有的3-ζ基组(Def2-TZVPP)能够在一定程度上“体现”弥散作用, 因此, 考察省略弥散函数对计算结果的影响.由图 9b可见, 在200~600 nm范围内, 弥散函数对MOA计算光谱的影响很小, 两条曲线几乎完全重合, 第一吸收峰位置相差波长 < 10 nm, 可以在计算中省略弥散函数.
 |
| 图 9 甲基橙紫外可见吸收光谱(pH=7, pH=12) (a为实验光谱(exp.), b为计算光谱(theo.)) Fig. 9 UV-vis absorption spectrum of methyl orange(pH=7, pH=12) (a is the experimental spectrum (exp.), and b is the calculated spectrum (theo.)) |
用pH=7的实验光谱作为对比, 分组讨论计算光谱与实验光谱(图 10).需要提前指出的是: 1)孤立簇模型的计算结果完全反映实验值的可能性较低, 因为分子运(振)动导致的构型在亚皮秒尺度不断波动(Xu et al., 2021), 实验光谱来自各种构型的权重贡献;2)体系中弱相互作用复杂, 理论模型在简化和抽象的过程中存在一定程度的妥协.图 10a表明MOA单体分子的紫外吸收光谱与实验光谱差距较大(463-394=69 nm), 暗示存在其他影响因素, MOAD在MOA的基础上有所进步, 更加贴近实验吸收光谱, 也较好地反映出第二吸收峰(图 10a中MOAD), MOAD团簇中发生了很明显的苯环平行错位(Grimme, 2008)和正负电势区域吸引现象, 表明MOA单体通过π-π叠加作用和静电引力结合, 彼此波函数互相扰动, 导致激发红移.SCMOA系列团簇由钠离子、MOA单体及水分子组成, 从峰位置角度看SCMOA_2和SCMOA_3表现最佳(图 10b ), 其预测的第一吸收峰位置约在424 nm处(实验值463 nm), SCMOA_2和SCMOA_3体现出钠离子对偶氮键的扰动, 由于孤对电子的存在, 偶氮键容易被阳离子极化, 根据第3.1节的讨论, 偶氮键的电子变化对吸收光谱存在显著影响, SCMOA_2和SCMOA_3模型还体现出实验光谱中的“肩峰”(约413 nm处), 两者计算光谱均存在类似结构.SCMOA_1团簇体现了钠离子和磺酸基的相互作用, 钠离子靠近可能会增强磺酸基的“吸”电子能力, 但由于本底水平已经较高, 因此扰动不明显, 第一吸收峰位置预测在396 nm附近, 表现不如SCMOA_2和SCMOA_3.
 |
| 图 10 MOA团簇结构的计算紫外可见吸收光谱 (exp.标识为实验光谱) Fig. 10 Theoretical UV-Vis spectrum of MOA cluster structures (exp. indicates an experimental spectrum) |
被寄予厚望的SCMOA_WC(第一激发峰预测在390 nm处)表现不佳, 不仅在SCMOA系列中表现最差(图 10b), 还在第一激发波长预测上逊色于MOA模型(图 10a中MOA, 第一激发峰在394 nm处).可能有两方面原因: 一方面, SCMOA_WC仅在与水分子高度亲和的磺酸基附近使用了显式水模型, 其他部分使用的是隐式水模型, 在一个分子尺度范围内两者耦合欠佳, 或许扩大显示水分子模型的作用范围能解决此问题(计算量剧增);另一方面, 钠离子的配位水分子削弱了其极化能力(Naζ+---Oζ-作用), 导致钠离子对偶氮键极化作用被削弱.但SCMOA_WC模型的真实性最好, 此部分工作会使用经典分子动力学和从头算分子动力学(AIMD)进一步深入研究.
SC2MOA系列模型真实存在的概率较小, 因为第一钠离子进入吸附位后扰动ESP导致第二处吸附位点削弱或消失, 但其结构能提供较为有用(有趣)的化学信息.SC2MOA_2模型中两个钠离子围绕偶氮键呈对称分布(图 8中SC2MOA_2, 图 10c), 显然两者极化作用也被“对称”了, 偶氮键波函数发生了对称极化, 导致其计算光谱得到了一个非常对称的“高斯”峰形, 失去了“肩峰”细节.SC2MOA_1具有“肩峰”特征, 表现出SCMOA_1、SCMOA_2的混合特征.上述研究表明, MOA分子的吸收光谱模型有待完善, 简单的MOA单体、二聚体不能很好地预测实验光谱, 钠离子配位的簇结构是较有潜力的模型.正确处理弱相互作用导致的偶氮键极化可能是正确预测实验光谱的关键.
4 结论(Conclusions)偶氮键在偶氮分子的紫外吸收中具有重要作用, 它并非激发电子的直接提供者, 而以“桥梁”身份沟通两侧π结构, 形成共轭降低激发能;偶氮苯分子实验光谱的第一吸收峰可指认为π→π*跃迁, 同时更低波长的n→π*跃迁禁阻;甲基橙分子的计算光谱不能简单地通过单体或二聚体反映, 溶液环境中其他因子与MOA的弱相互作用对紫外光谱存在显著影响, 二(多)聚体-钠离子-水分子组成的团簇是有潜力的预测模型.
致谢: 感谢孙浩、许欢、纪光晗同学的实验辅助工作.
Abraham M J, Murtola T, Schulz R, et al. 2015. GROMACS: High performance molecular simulations through multi-level parallelism from laptops to supercomputers[J]. SoftwareX, 1-2: 19-25. DOI:10.1016/j.softx.2015.06.001 |
Cai R Q, Zhang B G, Shi J X, et al. 2017. Rapid photocatalytic decolorization of methyl orange under visible light using VS4/carbon powder nanocomposites[J]. ACS Sustainable Chemistry & Engineering, 5(9): 7690-7699. |
Cattaneo P, Persico M. 1999. An abinitio study of the photochemistry of azobenzene[J]. Physical Chemistry Chemical Physics, 1(20): 4739-4743. DOI:10.1039/a905055h |
Döbbelin M, Ciesielski A, Haar S, et al. 2016. Light-enhanced liquid-phase exfoliation and current photoswitching in graphene-azobenzene composites[J]. Nature Communications, 7(1): 11090. DOI:10.1038/ncomms11090 |
Duarte L, Fausto R, Reva I. 2014. Structural and spectroscopic characterization of E- and Z-isomers of azobenzene[J]. Physical Chemistry Chemical Physics, 16(32): 16919-16930. DOI:10.1039/C4CP00240G |
Escudero D, Trupp S, Bussemer B, et al. 2011. Spectroscopic properties of azobenzene-based pH indicator dyes: A quantum chemical and experimental study[J]. Journal of Chemical Theory and Computation, 7(4): 1062-1072. DOI:10.1021/ct1007235 |
Frisch M J, Trucks G W, Schlegel H B, et al. 2009. Gaussian 09, Revision A. 02[CP]. Gaussian, Inc. : Wallingford, CT
|
Gagliardi L, Orlandi G, Bernardi F, et al. 2004. A theoretical study of the lowest electronic states of azobenzene: the role of torsion coordinate in the cis-trans photoisomerization[J]. Theoretical Chemistry Accounts, 111(2): 363-372. |
Goerigk L, Grimme S. 2010. Assessment of TD-DFT methods and of various spin scaled CIS(D) and CC2 versions for the treatment of low-lying valence excitations of large organic dyes[J]. The Journal of Chemical Physics, 132: 184103. DOI:10.1063/1.3418614 |
Grimme S, Ehrlich S, Goerigk L. 2011. Effect of the damping function in dispersion corrected density functional theory[J]. Journal of Computational Chemistry, 32(7): 1456-1465. DOI:10.1002/jcc.21759 |
Grimme S. 2008. Do special noncovalent π-π stacking interactions really exist?[J]. Angewandte Chemie International Edition, 47(18): 3430-3434. DOI:10.1002/anie.200705157 |
Guan R Q, Li J X, JZhang J K, et al. 2019. Photocatalytic performance and mechanistic research of ZnO/g-C3N4 on degradation of methyl orange[J]. ACS Omega, 4(24): 20742-20747. DOI:10.1021/acsomega.9b03129 |
Liu Y D, Yuan Y Z, Tian X H, et al. 2020a. Computational design of p-(dimethylamino)benzylidene-derived push-pull polyenes with high first-hyperpolarizabilities[J]. Physical Chemistry Chemical Physics, 22(9): 5090-5104. DOI:10.1039/C9CP05631A |
Liu Y D, Yuan Y Z, Tian X H, et al. 2020b. High first-hyperpolarizabilities of thiobarbituric acid derivative-based donor-π-acceptor nonlinear optical-phores: Multiple theoretical investigations of substituents and conjugated bridges effect[J]. International Journal of Quantum Chemistry, 120(10): 26176-26189. |
Liu Z Y, Lu T, Chen Q X. 2020. c.An sp-hybridized all-carboatomic ring, cyclo[18]carbon: electronic structure, electronic spectrum, and optical nonlinearity[J]. Carbon, 165: 461-467. DOI:10.1016/j.carbon.2020.05.023 |
Lu T, Chen F W. 2012. Multiwfn: a multifunctional wavefunction analyzer[J]. Journal of Computational Chemistry, 33(5): 580-592. DOI:10.1002/jcc.22885 |
Lu T, Chen Q X. 2020. A simple method of identifying π orbitals for non-planar systems and a protocol of studying π electronic structure[J]. Theoretical Chemistry Accounts, 139(2): 25-37. DOI:10.1007/s00214-019-2541-z |
Maier J A, Martinez C, Kasavajhala K, et al. 2015. FF14SB: Improving the accuracy of protein side chain and backbone parameters from FF99SB[J]. Journal of Chemical Theory and Computation, 11(8): 3696-3713. DOI:10.1021/acs.jctc.5b00255 |
Manzoni V, Modesto-Costa L, Nero J D, et al. 2019. Strong enhancement of NLO response of methyl orange dyes through solvent effects: A sequential Monte Carlo/DFT investigation[J]. Optical Materials, 94: 152-159. DOI:10.1016/j.optmat.2019.05.018 |
Marenich A V, Cramer C J, Truhlar D G. 2009. Universal solvation model based on solute electron density and on a continuum model of the solvent defined by the bulk dielectric constant and atomic surface tensions[J]. Journal of Physical Chemistry B, 113(18): 6378-6396. DOI:10.1021/jp810292n |
Martinez L, Andrade R, Birgin E G, et al. 2009. PACKMOL: A package for building initial configurations for molecular dynamics simulations[J]. Journal of Computational Chemistry, 30(13): 2157-2164. DOI:10.1002/jcc.21224 |
Petrella A, Petrella M, Boghetich G, et al. 2013. Laboratory scale unit for photocatalytic removal of organic micropollutants from water and wastewater.Methyl orange degradation[J]. Industrial & Engineering Chemistry Research, 52(6): 2201-2208. |
Samanta D, J, Chu Z L, et al. 2018. Reversible photoswitching of encapsulated azobenzenes in water[J]. Proceedings of the National Academy of Sciences, 115(38): 9379-9384. DOI:10.1073/pnas.1712787115 |
Schauperl M, Nerenberg P S, Jang H, et al. 2020. Non-bonded force field model with advanced restrained electrostatic potential charges (RESP2)[J]. Communications Chemistry, 3(1): 44. DOI:10.1038/s42004-020-0291-4 |
Schmider H L, Becke A D. 2000. Chemical content of the kinetic energy density[J]. Journal of Molecular Structure: THEOCHEM, 527(1): 51-61. |
Tang H R, Stanbury D M. 1994. Direct detection of aqueous diazene: Its UV spectrum and concerted dismutation[J]. Inorganic Chemistry, 33(7): 1388-1391. DOI:10.1021/ic00085a029 |
Titov E, Lysyakova L, Lomadze N, et al. 2015. Thermal cis-to-trans isomerization of azobenzene-containing molecules enhanced by gold nanoparticles: An experimental and theoretical study[J]. The Journal of Physical Chemistry C, 119(30): 17369-17377. DOI:10.1021/acs.jpcc.5b02473 |
童沈阳. 1980. 甲基橙在酸性溶液中的变色机理[J]. 化学通报, (3): 32-37. |
Walker M, Harvey A J A, Sen A, et al. 2013. Performance of M06, M06-2X, and M06-HF density functionals for conformationally flexible anionic clusters: M06 functionals perform better than B3LYP for a model system with dispersion and ionic hydrogen-bonding interactions[J]. The Journal of Physical Chemistry A, 117(47): 12590-12600. DOI:10.1021/jp408166m |
Wang J M, Wolf R M, Caldwell J W, et al. 2004. Development and testing of a general amber force field[J]. Journal of Computational Chemistry, 25(9): 1157-1174. DOI:10.1002/jcc.20035 |
Xu J H, Sun Z R, Zhang C Y, et al. 2021. Importance of nuclear quantum effects on the hydration of chloride ion[J]. Physical Review Materials, 5(1): L01280-1-L01280-6. DOI:10.1103/PhysRevMaterials.5.L012801 |
Yan M Q, Dryer D, Korshin G V. 2016. Spectroscopic characterization of changes of DOM deprotonation-protonation properties in water treatment processes[J]. Chemosphere, 148: 426-435. DOI:10.1016/j.chemosphere.2016.01.055 |
Yang Y, Wang G Z, Deng Q, et al. 2014. Microwave-assisted fabrication of nanoparticulate TiO2 microspheres for synergistic photocatalytic removal of Cr(VI) and methyl orange[J]. Acs Applied Materials & Interfaces, 6(4): 3008-3015. |
Yu X J, Wang Z B, Buchholz M, et al. 2015. cis-to-trans isomerization of azobenzene investigated by using thin films of metal-organic frameworks[J]. Physical Chemistry Chemical Physics, 17(35): 22721-22725. DOI:10.1039/C5CP03091A |
Zhang J H, Liu Q, Chen Y M, et al. 2012. Determination of acid dissociation constant of methyl red by multi-peaks gaussian fitting method based on UV-visible absorption spectrum[J]. Acta Physico-Chimica Sinica, 28(5): 1030-1036. DOI:10.3866/PKU.WHXB201203025 |
Zhao Q, Wang K W, Wang J L, et al. 2019. Cu2O nanoparticle hyper-cross-linked polymer composites for the visible-light photocatalytic degradation of methyl orange[J]. ACS Applied Nano Materials, 2(5): 2706-2712. DOI:10.1021/acsanm.9b00210 |