 2020, Vol. 40
2020, Vol. 40



2. 江苏省大气环境监测与污染控制高技术研究重点实验室, 南京 210044;
3. 北京大学环境科学与工程学院, 环境模拟与污染控制国家重点联合实验室, 北京 100871
2. Jiangsu Key Laboratory of Atmospheric Environment Monitoring and Pollution Control, Nanjing 210044;
3. State Key Joint Laboratory of Environmental Simulation and Pollution Control, College of Environmental Sciences and Engineering, Peking University, Beijing 100871
氨(NH3)在大气环境中广泛存在并扮演着重要的角色, 可以参与许多大气化学反应过程(唐孝炎等, 2006).氨气能参与到大气气溶胶的生成过程, 促进新粒子的生成(Ge et al., 2011; Kulmala et al., 2013).同时, 氨气作为碱性气体, 能够中和大气中的酸性气体, 进而改变大气化学性质;大气中氨/铵盐的气粒分配平衡可以显著改变颗粒物的酸碱度, 对大气中雾霾的生成机制有着至关重要的影响(Smith et al., 2010; Xie et al., 2017).检测大气中氨气的污染情况对于研究大气气溶胶的生成具有重要的意义, 同时也对氨气污染排放治理具有重要的参考价值.
大气中氨气的检测主要存在以下几个难点:大气中的氨气浓度差异较大, 浓度分布从几个ppbv到几百个ppbv(Sutton et al., 2001; Krupa, 2003), 实现对痕量氨气的检测具有一定的难度;氨在大气中以气态、颗粒态存在, 也可以水溶液的形式存在并可迅速在各个相态之间转换(唐孝炎等, 2006), 这使得气态氨的检测变得异常复杂, 易受干扰;而且气态氨分子具有较大的粘性, 易附着在采样装置和进样管路上, 产生记忆效应, 进而影响检测效率和仪器的检测响应时间(Yokelson et al., 2003).
近年来, 科研人员相继研发出一些大气中氨气的检测技术和方法, 目前应用于大气中氨气的检测方法主要可以分成离线和在线两类.离线检测的核心原理是用人工的方式将气态氨气从大气样品中收集、脱离或者吸收后收集存储(Keuken et al., 1988), 然后将收集到的样品采用溶解、浓缩或者稀释等方法预处理后进行化学或者仪器分析.目前常见的离线检测方法有纳氏分光光度法(王余萍等, 2008)、气相色谱法(GC)(傅若农等, 2003; 王绍臣等, 2016)、毛细管电泳法(CE)(Dabek-Zlotorzynska et al., 1998)等.这一类方法最主要的缺点是时间分辨率较低, 无法做到实时分析, 对大气中氨和有机胺短时间内的变化无法监测, 当需要较多样品进行分析时会消耗大量的人力和物力;此外, 在对样品的处理和制备过程, 以及化学分析过程中也可能出现人为污染的情况, 从而增加了结果的不确定性.在线检测仪器和方法主要有:在线环形吸收取样分析(AMANDA)系统(Wyers et al., 1993)、光腔衰荡光谱技术(CRDS)(Lundsbergnielsen et al., 1993; Martin et al., 2015)、开放光程-傅里叶红外光谱(OP-FTIR)法等(Schär et al., 1995; 徐亮等, 2007).近几年国外也有研究将质谱在线监测引入到大气中痕量气体的检测实验中(Sellegri et al., 2005a; Nowak et al., 2006; Hanson et al., 2011; Yu et al., 2012).其中, 质子转移反应质谱(PTR-MS)是在20世纪90年代由奥地利因斯布鲁克大学的Lindinger及其同事共同研发的(Lindinger et al, 1998; Yuan et al., 2017), PTR-MS能够实现在不对样品气体进行预处理的情况下实时在线地测量大气中的VOCs等多种气态物质, 并且具有高响应时间、高分辨率和高灵敏度的优势(Blake et al., 2009), 已经被多次应用于包括医学(Romano et al., 2015)、大气化学(Stockwell et al., 2015; Klein et al., 2016)、食品科学(Pedrotti et al., 2019)在内的许多科学领域.但在以往的研究中利用PTR-MS对大气中的氨气进行在线测量的研究较少, 例如, Sellegir等(2005a; 2005b)曾经在芬兰北部的林区使用一台PTR-CIMS观测了大气中的醛、醇、有机酸和氨等物质;Hanson等(2011)在美国亚特兰大部署了一台AmPMS用于监测氨气及C1~C5的气态有机胺;Zheng等(2015a)在中国南京使用一台高分辨率飞行时间化学电离质谱(HRToF-CIMS)监测了当地大气中的氨和有机胺.以上几组研究都是使用质子化的水(H3O+(H3O)n)作为质子反应的反应离子.Norman等(2007)曾将O2+作为反应离子, 使用一台PTR-MS检测了实验室条件下的氨气.Nowak等(2002; 2006)和Yu等(2012)用质子化的乙醇((C2H5OH)nH+)作为反应离子, 使用PTR-CIMS分别对氨进行检测.本研究拟采用质子化的丙酮((C3H6O)nH+)作为反应离子对大气中的氨进行观测.相比于使用质子化的水和乙醇作为反应离子, 质子化的丙酮((C3H6O)nH+)的质子亲和力更高(194.1 kcal · mol-1)(Yu et al., 2012), 对于碱基化合物的选择性更好, 能使氨气在不受大气中其他大量气态有机化合物干扰的情况下发生电离, 可有效避免其他物质对质谱检测结果的影响.
本研究观测站点位于山东省鲁西北平原的平原县农村地区, 与其他城市地区相比, 具有典型的农村生产生活排放特征, 更能代表我国北方农村地区的空气污染特征, 对该地大气中的氨进行系统观测和研究能够促进对该地区氨排放特征和排放来源的了解.因此, 本研究采用一台以质子化的丙酮离子((C3H6O)nH+)为反应离子的质子转移反应质谱仪(PTR-MS), 分别于2017年和2018年冬季在该地区对近地面大气中的氨进行长时间实时在线观测实验, 同时对当地大气中氨的污染特征和污染来源进行分析.
2 材料与方法(Materials and methods) 2.1 外场观测概况图 1是观测点所在的山东省德州市平原县部分地图.平原县地处山东省西北部, 属于黄河下游的鲁西北平原, 地势平坦, 气候属于半温润大陆性季风气候, 具有显著的大陆气候特征.图中的黄点标注的是观测仪器所在的德州市平原县国家气象局内的气象观测场(37.1°N, 116.4°E), 该站点南部为农业生产区, 覆盖有大片的农田和农村聚落(绿色框范围内), 北部分布有少量小型加工工业活动, 主要生产螺丝等小商品(蓝色框范围内).周边无高大建筑, 地势开阔, 适于大气污染的采样和观测研究.
 |
| 图 1 山东德州平原县观测点示意图 Fig. 1 Map of the sampling sites |
PTR-MS放置在一个能够保持室内恒温的可移动式观测方舱内, 方舱周边空旷, 距离最近的主要建筑物超过100 m, 在30 m范围内, 没有高过进样口的遮挡物, 观测场地周边被草地和低矮的灌木覆盖.观测时间分别为2017年11月9日—2018年1月10日、2018年11月12日—2019年1月2日.气体进样口被设置在观测方舱顶部, 大气样品通过一个外径1.9 cm、长2.5 m的聚四氟乙烯管进入质谱仪(PTR-MS).为了减少管壁损失, 采用大流量采样的方法, 利用气体泵加限流孔的方式将采样口的流量控制在10 L · min-1.
在观测实验期间, 除了使用一台PTR-MS对大气中的氨气进行实时在线观测以外, 还使用Thermo Scientific公司生产的Model 48i CO分析仪、Model 49i O3分析仪、Model 42i NOx分析仪、Model 43i SO2分析仪进行在线观测, 同时使用Megee公司生产的AE31型Aethalometer黑炭仪在880 nm波长下测定黑炭(BC), 数据的时间分辨率均为1 min.观测期间的气象数据, 包括温度、气压、风向、风速和相对湿度等则由观测点所在的平原县气象局气象观测中心提供, 时间分辨率为5 min.
2.2 PTR-MS仪器结构本研究使用一台自行搭建的质子转移反应质谱仪(PTR-MS), 并配备有电晕放电离子源(质子化的丙酮((C3H6O)nH+)离子源) (Fortner et al., 2004; Zheng et al., 2015a; 2015b; Ma et al., 2016).PTR-MS的结构如图 2所示, 具体由丙酮液罐、点到面的电晕放电(Corona Discharge)离子源、漂移管(Drift Tube)和四极杆质谱(Extrel, MAX 1000)组成(Ma et al., 2016).在使用质谱工作的过程中, 完全密闭的丙酮液罐在负压条件下变成丙酮蒸汽, 通过针阀控制流量, 同时将丙酮液罐放在冰水混合物罐内使得丙酮罐的温度维持在0 ℃, 以控制丙酮蒸汽压, 从而控制进入电晕放电离子源的气态丙酮能够维持恒定流量.电晕放电是在2200 V电压下两个电极之间形成电弧, 使丙酮气体电离产生反应离子(Zheng et al., 2015b; Ma et al., 2016).与传统的放射源放射产生的反应离子(Nowak et al., 2007; Yu and Lee, 2012)相比, 具有离子强度高、无放射性的优点.离子源产生的质子化的丙酮((C3H6O)nH+)通过一个直径为1 mm的小孔进入漂移管, 漂移管由11个不锈钢金属环用聚四氟乙烯(PTFE)垫片逐个隔开并连接在一个气密的腔体, 即离子漂移管(de Gouw et al., 2007).不锈钢金属环之间由1.0 MΩ的电阻器连接, 产生均匀电场, 从而控制离子漂移速率和离子分子反应时间(Yuan et al., 2017).同时通过控制漂移管内部压强, 使带电的质子化的丙酮((C3H6O)nH+)与空气中的中性分子(氮气和氧气等)碰撞分解, 减少反应离子团簇, 从而提高待测物质的离子产率和质谱检测效率.漂移管部分安装有安捷伦(Agilent Tris-300)干式卷轴泵, 通过该泵可以控制漂移管内气压在3.0 Torr (Hansel et al., 1995; Yu et al., 2012; Ma et al., 2016), 并由Agilent CDG-500电容薄膜压力计实时测得.在漂移管的外部安装有小功率加热套, 通过加热套将漂移管温度控制在40 ℃左右, 以避免高湿度下大气样品中的水气在管壁凝结而导致氨气损失, 又可以减少温度变化造成的氨气释放和正检测误差.经多次实验表明, 在40 ℃条件下, PTR-MS具有更好的检测灵敏度且不会因过热损坏漂移管结构.漂移管和后部的高真空部分之间由一个孔径为200 μm的限流孔隔离, 真空腔内部安装有四极杆质谱和电子倍增器, 由此检测待检测物质并获得信号值.真空腔的真空环境通过两个Agilent TV-301涡轮分子泵维持在1×10-5 Torr, 配置一台Agilent IDP-3干式卷轴泵作为两台分子泵的前置泵.实验中NH3通过与丙酮试剂离子发生质子转移反应而被检测.
 |
| 图 2 自主研发搭建的质子转移反应质谱(PTR-MS)示意图 Fig. 2 The schematic diagram of proton transfer reaction mass spectrometry (PTR-MS) |
PTR-MS的工作原理是质子转移反应(Lindinger et al., 1998).本研究中使用的离子源系统是一个自主设计安装的电晕放电离子源(Zheng et al., 2015a; 2015b), 以丙酮为工作液, 产生质子化的丙酮((C3H6O)nH+).带上电荷的丙酮会与进入到PTR-MS中的气态物质在漂移管(Drift Tube)中发生质子转移反应(Futrell, 1986).将质子化的丙酮作为反应离子时, 与氨气(NH3)的反应过程见式(1).在检测大气中的痕量物质时, 由于这些物质在大气中的浓度很低, 所需消耗的反应离子只占很少一部分, 不会造成反应离子的大量减少.
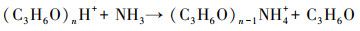
|
(1) |
大气中的部分痕量气体具有一定粘滞性, 如氨气(NH3)、硝酸(HNO3)等, 它们会附着在进样口和进样管路上, 产生记忆效应和气体损失, 从而造成检测结果的延迟和误差(Yokelson et al., 2003; Yu et al., 2012).之前的研究表明, 聚四氟材料(Teflon)对大气中氨气的吸附作用比较小, 适用于大气中痕量气体的采集(Yu et al., 2012).本研究选用总长为2.5 m、外径为1.9 cm的聚四氟乙烯管作为进样管路, 并使用采样泵加装限流孔的方式控制总采样流量维持在10 L · min-1, 以此来减少管壁损失, 提高检测结果的准确性.
质谱仪在运行过程中会产生一定的背景噪音信号(Zheng et al., 2015a), 为了除去背景噪音值对监测结果的影响, 本仪器配置了一个定制的零气发生装置(Zheng et al., 2015a; 2015b).在背景气路中, 将来自采样管路的气体通过一个350 ℃的铂催化转化炉, 可以将大气中的杂质气体在高温下分解, 产生干净无污染的零气再通入到检测仪器中, 而样气湿度不会变化(图 3).由于氨气本身的粘滞性, 导致背景气路的气体进入质谱仪后仍会有一定的氨气被检测到, 产生氨气的信号值, 而通过固定时间做背景的方式, 可以消除这种情况带来的误差, 提高检测结果的准确性.本次观测研究中, 使用一个电磁三通阀来控制采样气路和背景气路气流的切换, 从而达到去除仪器本身产生的背景噪音和氨气的粘滞性对检测结果影响的目的.样品信号和背景信号的检测时间分别为50 min和10 min, 对检测信号进行实时的矫正.
 |
| 图 3 仪器信号值图谱 Fig. 3 Instrument signal spectrum |
由质谱仪测得的数据需要进行归一化处理, 其目的是为了消除产物离子对反应离子浓度变化的敏感性而造成的信号变化.为了消除反应离子信号的变化对产物离子浓度变化的影响, 处理过程中将产物离子的信号值根据标准试剂离子的信号值(1×106 Hz)进行归一化, 并利用归一化后的信号值计算得到待测物质的浓度, 计算方法见式(2).
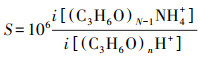
|
(2) |
式中, S为归一化后信号值, i[(C3H6O) nH+]和i[(C3H6O)N-1NH4+]分别为反应离子和产物离子的信号.
经过归一化后的信号值, 根据Zheng等(2015a)实验得到的标定系数校正后换算得到转换系数, 将氨气的信号值转换成浓度值.在下面的分析中将原始数据(85 s)平均到5 min后进行分析.
仪器的检测限可根据PTR-MS在背景路采样期间获得的背景信号值3倍标准偏差(3σ)计算得到, 本研究中根据背景信号计算得到的仪器检测限为0.03 ppbv.
3.2 PMF受体模型PMF即正交矩阵因子分解法(Positive Matrix Factorization), 是一种多元统计分析方法, 最早由Tapper和Paatero于1993年提出, 并由Paatero在1997年完善(Paatero, 1997).目前已经被广泛应用于大气环境悬浮颗粒物及挥发性有机物的源解析分析中(Amato et al., 2012; Yuan et al., 2012).PMF模型将多个样本、多物种的采样观测数据作为一个n×m的矩阵X (i, j), 其中i代表受体样本, j代表物种, 这样矩阵X可以分解为矩阵G和矩阵F.PMF模型的目标就是通过模拟计算找出p个污染源的成分谱矩阵 F (k, j)p×m和p个污染源在每一个样本中的贡献矩阵 G (i, k)n×p, k代表污染源.其中, 第ij个样本的观测浓度可以表示为:
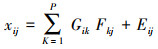
|
(3) |
式中, i、j、k、n、m、p均为正整数, xij为数据矩阵X中的一个浓度数据, 即第j个样品中的第i个物种浓度, Gik为源成分谱矩阵G中的元素, 即第k个来源中的第i个物质, Fkj为源贡献矩阵F中的第k个源对第j个样品的贡献, Eij为残差.通过PMF模型计算目标函数Q设置最优解:
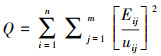
|
(4) |
式中, n为物种数, m为样品数, uij为xij的标准偏差, 在gik≥0, fik≥0的约束条件下, 通过对Q迭代最小化, 确定G和F.
PMF模型使用自展方法来估计解析结果的稳定性, 模拟计算出与采样获得的原始数据一致的另一组数据来分解出源谱图和源贡献矩阵, 并同原始采样数据进行比较, 使源解析结果更可信.与其它源解析方法相比, PMF模型不需要已知的源成分谱数据, 可利用数据标准差对结果进行优化, 分解矩阵中元素负担率为非负值, 并且在数据存在遗漏和缺失的情况下依然可以分析.目前多个课题和多地对PMF方法进行了验证和分析, 均表明PMF解析结果与检测结果接近, 准确度很高.
3.3 数据分析与结果本研究分别于2017年11月7日—2018年1月10日和2018年11月12日—2019年1月2日期间, 将PTR-MS部署于山东省德州市平原县气象局内的气象观测场进行外场观测实验.观测期间, 使用质子化的丙酮((C3H6O)nH+作为反应离子的PTR-MS稳定运行, 获得了长期、高分辨率的近地面氨气浓度数据.
图 4为两次观测期间获得的氨浓度数据和其他气象数据的时间序列.由图可知, 2017年冬季观测期间温度为-9.3~20.4 ℃, 平均温度为(1.9±5.3) ℃;相对湿度为12%~99%, 平均相对湿度为49.3%±22.6%;风速为0~9.4 m · s-1, 风向以东北和西南风为主.2018年冬季观测期间温度为-14.0~16.2 ℃, 平均温度为(1.6±6.2) ℃;大气相对湿度为14%~98%, 平均约为62.2%±22.7%, 整体上高于2017年冬季水平;风速为0~7.1 m · s-1, 风向以东北和西南风为主.在观测期间氨的浓度变化范围比较大, 从几个ppbv到几十个ppbv, 两次观测平均值分别为(5.89±5.27) ppbv和(2.65±2.41) ppbv, 其中, 2017年冬季观测期间氨浓度为0.048~71.68 ppbv, 在2017年12月10日上午6:00观测到最大值71.68 ppbv.2018年冬季观测期间氨浓度为0.14~21.22 ppbv, 在18年11月24日凌晨0:40观测到最大值21.22 ppbv.整体上看, 2018年冬季大气中的氨浓度有所降低, 这与当年气象状况的差异有一定的相关性.
 |
| 图 4 2017年(a)及2018年(b)冬季观测期间德州氨气和其他气象数据的时间序列 Fig. 4 Time series of ammonia and meteorological data during the 2017(a) and 2018(b) winter |
氨气在两个冬季观测期间均呈现出明显的日变化规律(图 5a、5b), 即在上午6:00—7:00左右出现峰值, 太阳升起后, 随着气温上升, 大气湿度下降, 氨气浓度开始下降, 下午约15:00出现一天当中的浓度低值, 随后上升.在观测点周边主要分布的是大量的农田和农村居民点, 且周边并没有大量化工企业排放氨气到大气中去.风玫瑰图(图 5c、5d)表明观测点周边污染物主要来自东北和西北方向, 这两个方向分布有大量的村落, 初步判断该地区近地面大气中的氨气可能主要由当地农村居民生活产生.调查表明, 当地村民有使用木柴生火做饭的习惯, 且在冬季有烧炕取暖的传统, 当夜晚来临时, 木柴、秸秆等生物质燃烧产生大量污染气体, 其中包括氨气被排放到大气中, 使得氨气浓度在夜间不断积累, 次日早晨由于边界层升高及日出后开始发生大气光化学反应, 氨气浓度开始下降, 并在下午约15:00达到最低值, 从而造成当地氨气浓度夜间高、日间低的情况.
 |
| 图 5 德州冬季氨气和气象数据的日变化情况及风玫瑰图(2017、2018年) Fig. 5 Averaged diurnal profiles of NH3, meteorological data and wind rose for Dezhou (2017and 2018) in winter |
在2017年冬季观测期间, 大气中氨浓度最大值为71.68 ppbv, 出现在12月10日上午6:00;并且在12月28日上午10:40出现氨气浓度在短时间内急剧升高的情况, 最高达到60.82 ppbv.这两个时间段都有着相似的气象条件, 即短时间内受到西北风向(图 6)的影响, 来自该方向的工厂生产活动排放的氨被PTR-MS捕获, 在短时间内形成很高的峰值.在这两个时间段内氨和CO的相关性显著(r分别为0.88和0.85), 而与NOx、O3、SO2等其他常规气体的相关不显著(r均小于0.5), 这可能与附近工业园区内生产活动造成的短时间排放有关.在2018年冬季观测期间, 11月23日夜间到11月24日凌晨, 氨气浓度在短时间内上升, 并在24日凌晨0:40达到观测期间最高值24.22 ppbv.在氨浓度升高的时间段内, 观测点主要受到西北风的影响(图 7), 与之前的情况相同, 氨与CO(r=0.88)、NO2(r=0.68)相关性显著, 但与其他物质相关性不显著.在2017年冬季和2018年冬季的3次氨气浓度短时间快速上升具有相同的特征, 即可能都受到西北部工业园排放的影响.在其他大多数观测时间段内, 观测点附近的氨主要来自周边农村的取暖等生活需求衍生的生物质和燃煤燃烧排放源在夜间的缓慢排放, 夜间排放的氨在大气中积累, 导致氨浓度升高.
 |
| 图 6 2017年冬季两次氨气浓度迅速升高时间段内的风玫瑰图 Fig. 6 Wind roses during the two periods when ammonia concentration rose rapidly in the winter of 2017 |
 |
| 图 7 2018年冬季氨气浓度迅速升高时间段内的风玫瑰图 Fig. 7 Wind roses during the periods when ammonia concentration rose rapidly in the winter of 2018 |
本研究使用美国国家环保部(USEPA)推荐的EPA-PMF 5.0, 将已经获得的氨气、CO、NOx、NOy、O3、SO2和BC数据导入软件进行源解析分析, 根据EPA-PMF 5.0使用说明, 对于由于维护仪器或者停电导致的缺测数据用-999替换.在PMF模型计算过程中, 将因子数尝试设置为2~10分别进行了100次迭代分析, 并删除其中坏点.最终在该观测点的两次观测中认定了4类因子, 根据每种因子的组分特征将它们识别为4种来源:周边农村夜间取暖排放(木材、煤炭等)、交通源排放、其他来源和外部传输源.
图 8分别给出了PMF解析的2017年和2018年冬季4类排放因子中不同化学组分的贡献占比, 柱形图表示不同污染物质在各个因子中的贡献百分比.在2017年冬季的PMF结果中, 因子1中氨、CO、SO2、BC的贡献较大.这与当地农村在冬季使用包括生物质燃料、小部分未经脱硫处理的煤等燃料取暖、生活做饭的排放特征相关联, 而BC的典型来源就是燃烧, 包括生物质燃烧和化石燃料燃烧.Tian等(2007)、Xie等(2007)和Li等(2016)研究了使用煤和生物质燃料(如木材、秸秆等)过程, Hegg等(1988)对木材燃烧过程中的氨排放进行了研究, 在这些燃烧过程中氨、CO、SO2会有明显的相关性.综上将因子1认定为周边农村取暖等生活燃料燃烧(木材、煤炭等).因子2中, 氨、CO、NO2、BC占比具有明显的相关特征.大量研究发现, 机动车尾气等交通源会释放氨(Cadle et al., 1980; 程刚等, 2016; 王凯等, 2018), Shelef等(1974)和Burgard等(2006)的研究指出, 氮氧化物和氢气的反应是汽车尾气中氨的主要来源, 汽车尾气排放伴随着NOx和CO等物质.因此, 将因子2认定为交通源排放.因子3中, 各个物质都有一定的占比, 但都较低, 没有明显的指示性, 将其认定为与观测站点附近的生产活动相关, 包括农田土壤释放、畜牧养殖排放、工业排放等在内的其他来源.因子4中, NOy和O3等二次污染物贡献占比较大, 符合大气的老化特征, 因此, 将因子4认定为外部远距离传输来源, 而且由于观测点远离大型都市、工业群落, 这部分来源对观测影响较小, 占比极少.
 |
| 图 8 2017年(a)及2018年(b)冬季各污染源因子的贡献率 Fig. 8 Contribution rates of various pollution sources in the winter of 2017(a) and 2018(b) |
通过因子的判定及各个污染源对氨的贡献率结果, 可以得出观测点测得的氨主要来自周边农村夜间排放、交通源排放.从图 9的贡献率可以看出, 氨的排放主要来自于周边农村取暖排放, 2018年冬季和2017年冬季占比分别为66.0%和55.0%, 交通排放源在两次观测中分别占到27.0%和36.8%, 其他周边排放源对该地区近地面大气中氨浓度的影响较小, 且由于氨的大气化学寿命短的特点, 外部传输来源极少(均小于1%).结合两次观测的氨气数据和PMF分析结果可以看出, 2018年冬季相比于2017年冬季, 大气中氨气总体浓度下降, 且2018年冬季来自周边农村冬季取暖排放的氨有较明显的减少, 交通源排放占比上升了9.8%.
 |
| 图 9 2017年和2018年冬季观测期间德州观测点大气中氨主要来源贡献率 Fig. 9 Relative contributions of ammonia sources for Dezhou (2017 and 2018) in winter |
在2017年和2018年冬季两次观测期间获得的氨浓度变化范围比较大, 从几个ppbv到几十个ppbv, 两次观测平均值分别为(5.89±5.27) ppbv和(2.65±2.41) ppbv.总体上2018年冬季观测点周边氨气浓度出现下降, 冬季大气中氨的浓度呈现出明显的日变化规律, 即在上午6:00—7:00左右出现峰值, 太阳升起后, 随着气温上升, 大气湿度下降, 氨气浓度开始下降, 下午约15:00出现一天当中的浓度低值, 随后上升.结合PMF分析可知, 德州观测点大气中氨气来源主要是周边农村在冬季大量使用生物质燃料燃烧取暖造成的生活排放和当地交通源排放, 两次冬季观测中当地农村取暖排放分别占到66.0%和55.0%, 交通排放源在两次观测中分别占到27.0%和36.8%.但来自观测点西北部的工业园区污染排放可能会因风向的变化造成观测点附近氨气在短时间内浓度迅速上升, 得益于PTR-MS对大气中氨的观测具有高时间分辨率的优势, 这些短时间、高浓度、危害较大的偶然源也可以被迅速识别.
在本次观测研究中, 对PTR-MS利用质子化的丙酮作为反应离子来检测大气中痕量氨的方法进行了实践和验证, 并获得了观测点的氨气浓度数据, 克服了大气中痕量氨气检测的难点.采用大流量采样的方法, 利用质子化的丙酮作为反应离子对碱性气体具有更好的选择性和检测准确性的优势.
Amato F, Hopke P K. 2012. Source apportionment of the ambient PM2.5 across St. Louis using constrained positive matrix factorization[J]. Atmospheric Environment, 46: 329-337. |
Blake R S, Monks P S, Ellis A M. 2009. Proton-transfer reaction mass spectrometry[J]. Chemical Reviews, 109(3): 861-896. DOI:10.1021/cr800364q |
Burgard D A, Bishop G A, Stedman D H. 2006. Remote sensing of ammonia and sulfur dioxide from on-road light duty vehicles[J]. Environmental Science & Technology, 40(22): 7018-7022. |
Cadle S H, Mulawa P A. 1980. Low-molecular-weight aliphatic amines in exhaust from catalyst-equipped cars[J]. Environmental Science & Technology, 14(6): 718-723. |
程刚, 李金香, 王欣, 等. 2016. 北京市交通环境春季大气氨污染水平分析[J]. 环境科学学报, 36(8): 2803-2810. |
Dabek-Zlotorzynska E, Maruszak W. 1998. Determination of dimethylamine and other low-molecular-mass amines using capillary electrophoresis with laser-induced fluorescence detection[J]. Journal of Chromatography B:Biomedical Sciences and Applications, 714(1): 77-85. DOI:10.1016/S0378-4347(98)00070-X |
De Gouw J, Warneke C. 2007. Measurements of volatile organic compounds in the earth's atmosphere using proton-transfer-reaction mass spectrometry[J]. Mass Spectrom Rev, 26(2): 223-257. DOI:10.1002/mas.20119 |
Fortner E C, Zhao J, Zhang R. 2004. Development of ion drift-chemical ionization mass spectrometry[J]. Analytical chemistry, 76(18): 5436-5440. |
傅若农. 2003. 国内气相色谱近年的进展[J]. 分析试验室, 22(2): 94-107. |
Futrell J H. 1986. Gaseous Ion Chemistry and Mass Spectrometry[M]. New York: Wiley.
|
Ge X, Wexler A S, Clegg S L. 2011. Atmospheric amines-Part I.A review[J]. Atmospheric Environmen, 45(3): 524-546. DOI:10.1016/j.atmosenv.2010.10.012 |
Hansel A, Jordan A, Holzinger R, et al. 1995. Proton transfer reaction mass spectrometry:on-line trace gas analysis at the ppb level[J]. International Journal of Mass Spectrometry and Ion Processes, 149: 609-619. |
Hanson D R, McMurry P H, Jiang J, et al. 2011. Ambient pressure proton transfer mass spectrometry:detection of amines and ammonia[J]. Environmental Science Technology, 45(20): 8881-8888. |
Hegg D A, Radke L F, Hobbs P V, et al. 1988. Ammonia emissions from biomass burning[J]. Geophysical Research Letters, 15(4): 335-337. |
Keuken M P, Schoonebeek C A M, Wensveen-Louter A V, et al. 1988. Simultaneous sampling of NH3, HNO3, HCl, SO2 and H2O2 in ambient air by a wet annular denuder system[J]. Atmospheric Environment, 22(11): 2541-2548. DOI:10.1016/0004-6981(88)90486-6 |
Klein F, Platt S M, Farren N J, et al. 2016. Characterization of gas-phase organics using proton transfer reaction time-of-flight mass spectrometry:Cooking emissions[J]. Environmental Science Technology, 50(3): 1243-1250. DOI:10.1021/acs.est.5b04618 |
Krupa S V. 2003. Effects of atmospheric ammonia (NH3) on terrestrial vegetation:a review[J]. Environmental Pollution, 124(2): 179-221. DOI:10.1016/S0269-7491(02)00434-7 |
Kulmala M, Kontkanen J, Junninen H, et al. 2013. Direct observations of atmospheric aerosol nucleation[J]. Science, 339(6122): 943-946. DOI:10.1126/science.1227385 |
Li Q, Jiang J, Cai S, et al. 2016. Gaseous ammonia emissions from coal and biomass combustion in household stoves with different combustion efficiencies[J]. Environmental Science & Technology Letters, 3(3): 98-103. |
Lindinger W, Jordan A. 1998. Proton-transfer-reaction mass spectrometry (PTR-MS):on-line monitoring of volatile organic compounds at pptv levels[J]. Chemical Society Reviews, 27(5): 347-375. |
Lundsbergnielsen L, Hegelund F, Nicolaisen F. 1993. Analysis of the high-resolution spectrum of ammonia (14NH3) in the near-infrared region, 6400-6900cm-1[J]. Journal of Molecular Spectroscopy, 162(1): 230-245. |
Ma Y, Diao Y, Zhang B, et al. 2016. Detection of formaldehyde emissions from an industrial zone in the Yangtze River Delta region of China using a proton transfer reaction ion-drift chemical ionization mass spectrometer[J]. Atmospheric Measurement Techniques, 9(12): 6101-6116. DOI:10.5194/amt-9-6101-2016 |
Martin N, Ferracci V, Cassidy N, et al. 2005. Cavity ring-down spectrometer (CRDS) development for ambient measurements of ammonia[J]. AGUFM, 2015: A31F-08. |
Norman M, Hansel A, Wisthaler A. 2007. O2+ as reagent ion in the PTR-MS instrument:Detection of gas-phase ammonia[J]. International Journal of Mass Spectrometry, 265: 382-387. DOI:10.1016/j.ijms.2007.06.010 |
Nowak J B, Huey L G, Eisele F L, et al. 2002. Chemical ionization mass spectrometry technique for detection of dimethylsulfoxide and ammonia[J]. Journal of Geophysical Research, 107(D18): 4346. |
Nowak J B, Huey L G, Russell A G, et al. 2006. Analysis of urban gas phase ammonia measurements from the 2002 Atlanta Aerosol Nucleation and Real-Time Characterization Experiment(ANARChE)[J]. Journal of Geophysical Research:Atmospheres, 111(D17308). DOI:10.1029/2006JD007113 |
Nowak J B, Neuman J A, Kozai K, et al. 2007. A chemical ionization mass spectrometry technique for airborne measurements of ammonia[J]. Journal of Geophysical Research:Atmospheres,, 112(D10S02). DOI:10.1029/2006JD007589 |
Paatero P. 1997. Least squares formulation of robust non-negative factor analysis[J]. Chemometrics and Intelligent Laboratory Systems, 37(1): 23-35. |
Pedrotti M, Khomenko I, Cappellin L, et al.2017. Implementation of rapid and non-invasive quality control through PTR-TOF-MS for anhydrous milk fat quality control[C].3rd IMEKOFOODS International Conference: Metrology Promoting Standardization and Harmonization in Food and Nutrition. GR
|
Romano A, Capozzi V, Spano G, et al. 2015. Proton transfer reaction-mass spectrometry:Online and rapid determination of volatile organic compounds of microbial origin[J]. Applied Microbiology Biotechnology, 99(9): 3787-3795. |
Schär K, Haus R, Heland J, et al. 1995. Measurements of atmospheric trace gases by emission and absorption spectroscopy with FTIR[J]. Berichte der Bunsengesellschaft für Physikalische Chemie, 99(3): 405-411. DOI:10.1002/bbpc.19950990325 |
Sellegri K, Hanke M, Umann B, et al. 2005. Measurements of organic gases during aerosol formation events in the boreal forest atmosphere during QUEST[J]. Atmospheric Chemistry and Physics, 5(2): 373-384. |
Sellegri K, Umann B, Hanke M, et al. 2005. Deployment of a ground-based CIMS apparatus for the detection of organic gases in the boreal forest during the QUEST campaign[J]. Atmospheric Chemistry and Physics, 5(2): 357-372. DOI:10.5194/acp-5-357-2005 |
Shelef M, Gandhi H. 1974. Ammonia formation in the catalytic reduction of nitric oxide.Ⅲ.The role of water gas shift, reduction by hydrocarbons, and steam reforming[J]. Industrial & Engineering Chemistry Product Research and Development, 13(1): 80-85. |
Smith J N, Barsanti K C, Friedli H R, et al. 2010. Observations of aminium salts in atmospheric nanoparticles and possible climatic implications[J]. Proc Natl Acad Sci USA, 107(15): 6634-6639. DOI:10.1073/pnas.0912127107 |
Stockwell C E, Veres P R, Williams J, et al. 2015. Characterization of biomass burning emissions from cooking fires, peat, crop residue, and other fuels with high-resolution proton-transfer-reaction time-of-flight mass spectrometry[J]. Atmospheric Chemistry and Physics, 15(2): 845-865. DOI:10.5194/acp-15-845-2015 |
Sutton M A, Tang Y S, Dragosits U, et al. 2001. A spatial analysis of atmospheric ammonia and ammonium in the U.K[J]. Scientific World Journal, 1(S2): 275-286. |
唐孝炎, 张远航, 邵敏. 2006. 大气环境化学(第2版)[M]. 北京: 高等教育出版社.
|
Tian F J, Yu J, McKenzie L J, et al. 2007. Conversion of fuel-N into HCN and NH3 during the pyrolysis and gasification in steam:a comparative study of coal and biomass[J]. Energy & Fuels, 21(2): 517-521. |
王凯, 樊守彬, 郭津津, 等. 2018. 北京市机动车尾气中氨气排放特征研究[J]. 环境工程, 36(3): 98-101. |
王绍臣. 2016. 气相色谱技术的研究进展及其应用[J]. 黑龙江科技信息, 22: 120-120. |
王余萍, 李永芳, 王冬梅. 2008. 纳氏试剂分光光度法测定空气氨方法探究[J]. 中国卫生检验杂志, 18(10): 2025-2026. |
Wyers G, Otjes R, Slanina J. 1993. A continuous-flow denuder for the measurement of ambient concentrations and surface-exchange fluxes of ammonia[J]. Atmospheric Environment Part A General Topics, 27(13): 2085-2090. DOI:10.1016/0960-1686(93)90280-C |
Xie H B, Elm J, Halonen R, et al. 2017. Atmospheric Fate of Monoethanolamine:Enhancing New Particle Formation of Sulfuric Acid as an Important Removal Process[J]. Environmental Science Technology, 51(15): 8422-8431. |
Xie J J, Yang X M, Zhang L, et al. 2007. Emissions of SO2, NO and N2O in a circulating fluidized bed combustor during co-firing coal and biomass[J]. Journal of Environmental Sciences, 19(1): 109-116. |
徐亮, 刘建国, 高闽光, 等. 2007. 开放光程傅里叶变换红外光谱系统观测城市空气中的NH3[J]. 大气与环境光学学报, 2(1): 60-63. |
Yokelson R J, Christian T J, Bertschi I T, et al. 2003. Evaluation of adsorption effects on measurements of ammonia, acetic acid, and methanol[J]. Journal of Geophysical Research, 108(D20): 4649. |
Yu H, Lee S H. 2012. Chemical ionisation mass spectrometry for the measurement of atmospheric amines[J]. Environmental Chemistry, 9(3): 190-200. |
Yuan B, Koss A R, Warneke C, et al. 2017. Proton-transfer-reaction mass spectrometry:Applications in atmospheric sciences[J]. Chemical Reviews, 117(21): 13187-13229. |
Yuan B, Shao M, de Gouw J, et al. 2012. Volatile organic compounds (VOCs) in urban air:How chemistry affects the interpretation of positive matrix factorization (PMF) analysis[J]. Journal of Geophysical Research:Atmospheres, 117(D24). DOI:10.1029/2012JD018236 |
Zheng J, Ma Y, Chen M, et al. 2015. Measurement of atmospheric amines and ammonia using the high resolution time-of-flight chemical ionization mass spectrometry[J]. Atmospheric Environment, 102: 249-259. |
Zheng J, Yang D, Ma Y, et al. 2015. Development of a new corona discharge based ion source for high resolution time-of-flight chemical ionization mass spectrometer to measure gaseous H2SO4 and aerosol sulfate[J]. Atmospheric Environment, 119: 167-173. DOI:10.1016/j.atmosenv.2015.08.028 |