 2020, Vol. 40
2020, Vol. 40



2. 粤港澳环境质量协同创新联合实验室, 广州 511443;
3. 齐鲁工业大学环境科学与工程学院, 济南 250353;
4. 中国科学院广州地球化学研究所, 有机地球化学国家重点实验室, 广州 510640;
5. 广东省环境监测中心, 广州 510308
2. Guangdong-Hongkong-Macau Joint Laboratory of Collaborative Innovation for Environmental Quality, Guangzhou 511443;
3. School of Environmental Science and Engineering, Qilu University of Technology, Jinan 250353;
4. The State Key Laboratory of Organic Geochemistry, Guangzhou Institute of Geochemistry, Chinese Academy of Sciences, Guangzhou 510640;
5. Guangdong Environmental Monitoring Center, Guangzhou 510308
近年来, 高浓度近地面臭氧(O3)以及大气细颗粒物(PM2.5)污染已成为我国许多地区日趋严峻的大气环境问题(Shao et al., 2009; Guo et al., 2014; 郑君瑜等, 2019).大气中的挥发性有机物(VOCs)是生成近地面O3和PM2.5的关键重要前体物(Seinfeld et al., 2006; 吕子峰等, 2009; 邓雪娇等, 2010; 王扶潘等, 2014), 影响大气环境和全球气候的同时, 也对人体健康造成一定的危害(胡珊等, 2009; Kassomenos et al., 2013).
深刻认识不同VOCs物种的源汇过程, 即来源和去除途径, 是准确评估VOCs对二次污染物贡献和开展VOCs防控的前提.VOCs主要来源于人为源和天然源释放(Williams, 2004).研究表明, 我国VOCs的人为源主要是交通运输、溶剂使用和生物质燃烧等过程.相关研究表明(Bo et al., 2008; Wei et al., 2008)天然源主要由植被排放所贡献, 从全球尺度上来说, 异戊二烯、单萜烯等是最具代表性的天然源VOCs物种, 可贡献天然源VOCs的90%以上(Guenther et al., 1995; Guenther et al., 2012; 李玲玉等, 2019).VOCs的去除一般可以通过发生化学反应或转化生成更高氧化态的物种(唐孝炎等, 2006), 主要化学去除途径是与OH自由基、NO3自由基、O3等发生反应(Atkinson et al., 2003).此外, 部分OVOCs物种尤其是低碳的醛酮化合物等自身会在光照条件下发生光解反应, 也为大气光化学反应提供了许多活性自由基.比如甲醛的光解是大气中HO2自由基重要的直接来源(Solberg et al., 2001; 尹鸿鸣等, 2008).日间, VOCs与OH自由基的氧化是最重要的化学去除途径(唐孝炎等, 2006), VOCs物种被OH自由基氧化去除的贡献比例最高可以达到98%以上(Ehhalt, 1999).而夜间NO3自由基对VOCs的氧化就显得更加重要, 最高也可占降解速率的88%以上(Li et al., 2018).
现有研究多数集中在OH自由基的氧化去除贡献, 而评估不同地区其他化学去除途径贡献的研究较少, 且大多数没有考虑OVOCs的光解.烷烃、芳香烃以及部分烯烃最主要的化学去除途径是OH自由基的氧化, OH自由基的氧化贡献VOCs去除速率可达到55%±16% (Geyer et al., 2001).但对于高活性的烯烃类物质, 其夜间与NO3自由基以及O3的氧化去除的贡献十分显著(de Gouw et al., 2017).此外, 典型的OVOCs(如丙酮)的光解最高可贡献其总化学去除速率的44% (Yuan et al., 2013).在我国, 华北区域大气氧化能力是华南地区的1.5倍, 两地均以OH自由基的氧化为主导, 华南与华北地区次要贡献去除途径则分别是NO3自由基和O3的氧化去除过程(栗泽苑, 2018).大多数现有研究仅针对当地VOCs的化学去除途径进行评估, 且评估的VOCs物种有限, 对不同地区(城市与区域)的对比还比较少.
珠江三角洲地区经济发展迅速, VOCs和其他污染物排放强度大, 且地处亚热带地区, 大气化学反应剧烈(Hofzumahaus et al., 2009; Lu et al., 2012), 导致了以O3污染为主的大气复合污染问题.珠三角地区已有大量研究开展了VOCs的测量与来源解析工作(Lau et al., 2010; Ling et al., 2011; Wu et al., 2016), 但对VOCs去除途径的研究还鲜有报道.本文将基于珠三角地区的城市站点和区域站点的两次外场观测数据, 针对不同VOCs组分的不同化学去除途径, 研究它们的去除速率及其贡献, 并分别对城市和区域站点人为源和天然源VOCs的化学去除途径进行去除速率贡献的分析, 深入理解不同VOCs物种的化学去除途径, 从而更好的认识VOCs在大气中的化学转化过程及对大气二次污染的贡献.
2 实验方法(Methods) 2.1 外场观测和测量方法本研究使用在珠三角地区开展的两次外场观测数据.城市观测站点布设在广州市天河区的中国科学院广州地球化学研究所(23.1°N, 113.2°E), 采样点距离地面高度约为25 m, 观测时间为2018年秋季(9—11月).区域观测的站点设在广东省江门市鹤山市桃源镇花果山的广东省大气环境超级监测站(22.7°N, 112.9°E), 站点处于广州和佛山两市的下风向, 是珠三角地区典型的区域受体站点(陈多宏等, 2012; 何俊杰等, 2013; 区宇波等, 2013).区域站点观测时间为2019年秋季(10—11月).
针对VOCs的测量, 两次外场观测使用了在线气相色谱质谱联用仪(GC-MS/FID)定量烷烃、烯烃、芳香烃等非甲烷烃类(NMHCs), 观测所使用的仪器物种检测限为10×10-12 ~84×10-12 mol · mol-1, 精密度为0.65%~9.14%.仪器运行期间, 每天利用包含56种VOCs组分的PAMS标气进行日校准, 观测中标气组分各物种的响应变化在±20%以内.同时, 两次观测也使用在线质子转移反应飞行时间质谱(PTR-TOF-MS)定量芳香烃和OVOCs等.PTR-TOF-MS具有高时间分辨率, 高灵敏度, 检测限较低等特点, 已广泛应用于外场观测(de Gouw et al., 2007; Yuan et al., 2017).观测所使用的仪器物种检测限为5×10-12~40×10-12 mol · mol-1, 精密度为6.10%~18.4%.仪器运行期间, 每天利用包含24种VOCs组分的钢瓶混合标气同时在干燥(RH < 1%)条件和大气湿度条件下进行标定校正, 以保证仪器运行期间数据的准确性.两次观测中, 标气组分响应因子波动范围均在20%以内.
同时, 对同期的常规痕量气体及气象参数进行了连续在线的监测.在两次观测中, O3、CO、SO2和NO的观测仪器分别为Thermo公司生产的O3分析仪(型号:49i)、增强型痕量CO分析仪(型号:48i-TLE)、增强型痕量SO2分析仪(型号:43i-TLE)、痕量NOx分析仪(型号:42i-TL).对于NO2的测量, 广州地化所中使用了Thermo 42i-TL痕量NOx分析仪, 而鹤山观测中使用了美国2B Technologies公司的Model 405 nm NO-NO2-NOx分析仪(Birks et al., 2018).对于颗粒物粒径分布的测量, 使用了TSI公司的扫描式迁移性粒径分析仪(SMPS, 10~650 nm)及空气动力学粒径谱仪(APS, 500~20000 nm).在鹤山观测中, 一套加湿浊度计系统(浊度计型号:EcoTech Aurora 3000)用于观测干燥状态下和80%相对湿度下的气溶胶散射系数和后续散射系数.光解速率常数的测量使用PFS-100光解光谱仪, 能够输出H2O2、HCHO、HONO、NO2、NO3和O1D等光解速率.气温、气压、相对湿度、风速、风向和降雨等气象参数则采用了Vaisala WXT520超声波气象传感器及Davis Vintage Pro 2气象站的数据.
2.2 利用模型模拟计算OH自由基及NO3自由基浓度在这两次观测中, OH自由基及NO3自由基的浓度均没有直接测量.本研究采用零维盒子模式Framework for 0-D Atmospheric Modeling (F0AM) v3.1 (Wolfe et al., 2016)对自由基浓度进行模拟, 用于评估VOCs物种不同去除途径的贡献.模型限制条件为实测的大气温度、压力、相对湿度、光解速率、常规气体(CO、NOx、O3等)和VOCs浓度数据.使用的化学机理为准详细化学反应机理(MCM v3.3.1) (Jenkin et al., 2012; Jenkin et al., 2015), 并增加了N2O5在颗粒物表面的吸附反应, 增加的反应速率常数数据来源于文献报道(Riedel et al., 2014; Wang et al., 2018a).在低NO浓度条件下, N2O5的非均相摄取反应是NO3自由基的一个较为重要的汇, 被添加至盒子模型中.
基于加湿浊度计系统的观测数据和环境相对湿度, 可计算出环境气溶胶含水量(Kuang et al., 2018)和环境状态下气溶胶表面积Saerosol (Kuang et al., 2019).该反应速率kN2O5是根据环境状态下气溶胶表面积Saerosol、N2O5的平均分子速度C, 以及摄取系数γN2O5, 依据式(1)计算得到的.模型中摄取系数γN2O5设置为0.002 (Wang et al., 2018a).
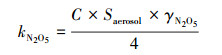
|
(1) |
盒子模型计算步长设置为1 h, 弛豫时间为3 d (Kumar et al., 2018), 对所有物种假定了寿命为24 h的损失过程, 以避免长寿命物种的浓度出现不合理的累积(Lu et al., 2012).
2.3 OVOCs光解速率常数计算在OVOCs的去除途径分析中, 还应考虑光解对醛酮类OVOCs的贡献.由于在观测期间, 光解光谱仪没有输出甲醛以外的OVOCs物种的光解速率常数, 故考虑结合各OVOCs物种光解速率常数随太阳天顶角(Solar Zenith Angle, SZA)变化的参数化公式计算得出各OVOCs的光解速率常数J (Saunders et al., 2003; Wolfe et al., 2016):

|
(2) |
在以上公式中, 针对每一个光解反应都有唯一的I、m、n 3个参数.这3个参数是通过45°N及高度为0.5 km的晴朗天空条件下的光化通量、吸收截面及量子产率通过最小二乘法拟合得到的(Jenkin et al., 1997; Saunders et al., 2003).本课题组在模型中需要通过实测的J(NO2)和计算的J(NO2)得到的比值对计算出的OVOCs光解速率进行校正, 以考虑云层遮挡等因素对光解速率的影响.经计算, 根据此方法估算的甲醛光解速率常数与光解光谱仪输出结果也比较吻合(R=0.998, 计算/实际的斜率为1.1296).
2.4 VOCs去除速率计算VOCs被各种氧化剂氧化消耗的反应速率一般是通过各VOCs物种的浓度、氧化剂的浓度和各氧化剂与VOCs的反应速率常数计算得出(Warneke et al., 2004):
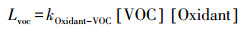
|
(3) |
而对于OVOCs如醛酮类物质, 其自身的光解也在该物质去除途径中占有一定比例, 因此对于VOCs物质总的去除速率Lvoc (10-12 · h-1)为:
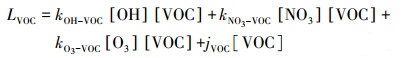
|
(4) |
式中, kOH-VOC、kNO3-VOC和kO3-VOC分别为OH自由基、NO3自由基、O3与VOCs物种的反应速率常数(Atkinson et al., 1992; Atkinson, 1997; Atkinson et al., 2003; Atkinson et al., 2006; Salgado et al., 2008; Keyte et al., 2013)以及IUPAC和JPL数据库中的数据(Burkholder et al., 2015).反应速率常数为文献值, 不确定度通常在20%以内(Atkinson et al., 2006), 但本文考虑不同化学去除途径的贡献, 该不确定度不会影响本文的结论. OH、NO3、O3和[VOC]分别为OH自由基、NO3自由基、O3和VOCs物种的浓度, jVOC为VOCs的光解速率常数.
根据盒子模式计算结果表明, 两次观测期间, OH自由基和NO3自由基浓度的日变化特征非常明显, 最高值分别出现在12:00和18:00.城市站点OH自由基的全天平均浓度为1.31×106 molecule · cm-3, 平均全天最高浓度为4.65×106 molecule · cm-3;区域站点则是6.15×105 molecule · cm-3和2.42×106 molecule · cm-3.NO3自由基在城市站点及区域站点的全天平均浓度分别为0.67×10-12 mol · mol-1和5.73×10-12 mol · mol-1, 最高浓度则是2.01×10-12 mol · mol-1和28.8×10-12 mol · mol-1, 这与国内典型大气环境下的NO3自由基浓度水平相符(Wang et al., 2017; Wang et al., 2018b).盒子模式对自由基的模拟结果的不确定度约为40%, 但由于计算得到的自由基浓度与文献值可比, 而且对于关键物种去除途径的结果与文献报道也较为一致, 认为OH自由基和NO3自由基的模拟对本文的结果影响不大.造成两地自由基浓度水平差异的原因之一是区域站点的NO较低, 使得NO + NO3 = NO2(k=2.6×10-11 cm3 · molecule-1 · s-1)的反应减弱, NO3自由基浓度得以累积, 出现比城市站点高的浓度(Sander et al., 2011).
3 结果与讨论(Results and discussion) 3.1 典型VOCs物种日变化分析典型VOCs物种在城市与区域站点的日变化如图 1所示.乙烷、乙烯、间+对-二甲苯作为典型的烃类物种, 主要来自一次人为源排放, 它们的日变化(图 1a~1c)均具有明显的夜间高、日间低的特点.受光化学消耗的影响, 其通常在午后15:00左右达到最低值, 随着光化学作用减弱, 一次排放增加, 浓度开始上升.很明显的看到, 乙烯和间+对-二甲苯与OH自由基反应更快, 活性更高, 因此在白天下午浓度水平的下降幅度更大.
 |
| 图 1 城市站点及区域站点乙烷(a)、间+对-二甲苯(b)、乙烯(c)、甲醛(d)、异戊二烯(e)与单萜烯(f)日变化特征 Fig. 1 Characteristics of diurnal variations for ethane (a), m, p-xylene (b), ethane (c), formaldehyde (d), isoprene (e) and monoterpenes (f) at the urban and regional sites |
异戊二烯和单萜烯作为天然源主要排放的烯烃, 它们的排放量还受到温度和光照的影响.如图 1e所示, 异戊二烯夜间浓度低、日间浓度高, 且最高值基本是出现在正午13:00左右, 这是由于它在夜间基本没有排放, 且日间光合作用的排放速率要远大于其化学去除的速率.单萜烯(图 1f)呈现白天低, 夜间高的相反趋势, 在夜间主要受边界层降低的影响而不断积累, 在早上7:00左右达到峰值, 之后随着光化学反应的发生, 浓度开始下降.而城市与区域站点夜间的差异可能是由于单萜烯与NO3自由基的反应在区域站点有较大的贡献.此外, 它们的浓度变化也会受到人为源排放的影响(Hellén et al., 2012).
甲醛作为典型的OVOCs, 其浓度变化受到了源排放、二次生成、光化学消耗和气象因素等各种作用的影响.如图 1d所示, 其日变化呈现了日间高, 夜间低的特征.日间随着边界层的上升和光化学反应的发生, 导致了甲醛的二次生成从而使其浓度上升, 在午后14:00左右达到峰值;之后随着二次生成速率逐渐低于其与OH自由基的氧化反应消耗速率, 其浓度开始下降.
3.2 不同种类VOCs组分化学去除途径分析基于VOCs去除速率计算方法, 本课题组分别计算了城市站点和区域站点观测期间, 上述典型VOCs物种被OH自由基、NO3自由基和O3平均去除速率的日变化.对于乙烷和间+对-二甲苯, 其最主要的去除途径是与OH自由基的氧化反应.如图 2a~2d所示, 它们与OH自由基的反应占其总去除速率的比例在两次观测中都达到了99%以上.
 |
| 图 2 城市站点及区域站点乙烷(a, b)、间+对-二甲苯(c, d)、乙烯(e, f)、异戊二烯(g, h)与单萜烯(i, j)在大气中被OH、NO3和O3氧化去除的平均去除速率 Fig. 2 Average loss rates by OH, NO3, and O3 oxidation for ethane (a, b), m, p-xylene (c, d), ethane (e, f), isoprene (g, h) and monoterpenes (i, j) at the urban and regional sites |
因乙烯分子中具有双键结构, 可与O3发生反应, 图 2e和2f为其在两次观测的总去除速率, 发现O3的氧化分别在城市站点和区域站点贡献了全天去除速率的10.9%和22.9%.区域站点的O3浓度要高于城市站点, 这也导致了两地O3氧化对去除速率贡献的差异.异戊二烯和单萜烯作为典型的天然源排放的烯烃, 各去除途径在两次观测的去除速率如图 2g~2j所示.NO3自由基的氧化对物种去除速率的贡献逐渐变得重要, 这是因为它们与NO3的反应速率常数相比于其他物种要大得多(Brown et al., 2012).同时, 与NO3自由基的氧化已成为单萜烯最重要的去除途径(67.6%).这也与Yuan等(2013)发现NO3自由基的氧化在2011年长岛观测中贡献了单萜烯去除速率的78.9%的研究结论非常相似.
两次观测中典型OVOCs物质各反应途径对其去除速率的贡献如图 3所示.不同于其他烃类物种, 甲醛、乙醛和丙酮等醛酮类化合物的光解对其去除有不可忽略的贡献.甲醛在城市站点占总去除速率的全天平均值的50.9%, 在区域站点中更是达到68.6%, 这与之前的研究结果吻合(Feilberg et al., 2007).而对于图 3中其他OVOCs物质, OH自由基的氧化去除仍然是最重要的去除途径, 如乙醛的总去除速率中, 光解分别在城市和区域站点占3.2%和6.5%.酮类化合物的光解速率尽管很小, 但对总去除速率的贡献也不小, 如丙酮的光解在城市和区域站点分别占23.9%和38.7%.
 |
| 图 3 城市站点及区域站点甲醛(a, b)、乙醛(c, d)、丙酮(e, f)在大气中被OH、NO3和O3氧化去除以及光解的平均去除速率 Fig. 3 Average loss rates by OH, NO3, O3 oxidation and photolysis for formaldehyde (a, b), acetaldehyde (c, d), and acetone (e, f) at the urban and regional sites |
本次观测中重点关注的烷烃、烯烃、芳香烃等烃类和典型的醇类、醛类、酮类等OVOCs的不同反应途径在整个观测期间的平均贡献比例汇总在图 4中.结果显示, OH自由基在大多数烷烃和芳香烃类物种的化学去除中占主导贡献, 除正构直链烷烃之外, 环烷烃与支链烷烃的主要去除途径也是与OH自由基反应.而烯烃与NO3自由基和O3的氧化在一定程度上显得尤为重要, 两者去除速率贡献最高能达到80%以上.需要特别指出的是, 苯乙烯作为取代基含有双键的芳香烃, 与NO3自由基具有较高的反应速率(1.5×10-12 cm3 · molecule-1 · s-1) (Atkinson et al., 2003), 在区域站点与NO3自由基的反应对去除速率的贡献可达77.9%.MVK和MACR作为异戊二烯的氧化产物, 其主要与OH自由基发生氧化反应而去除, 但在区域站点, 其与O3的氧化也有15.2%的贡献.而芳香烃的氧化产物苯酚和甲酚因与NO3自由基的反应速率较高, 分别达到了3.8×10-12 cm3 · molecule-1 · s-1和1.4×10-11 cm3 · molecule-1 · s-1 (Atkinson et al., 1992), 因此其与NO3自由基的反应是其最重要的去除途径, 在区域站点贡献均达到95%以上.
 |
| 图 4 城市站点(a)及区域站点(b)不同烃类及OVOCs在大气中被OH、NO3和O3氧化去除以及光解的去除速率贡献比例 Fig. 4 Relative importance of loss rates for all of hydrocarbons and OVOCs measured at the urban site (a) and regional site (b). Note that 2-methylpentane, n-dodecane and cis-2-butene were not measured at the regional site |
人为源代表性VOCs, 如苯、甲苯、乙烷、乙烯等, 主要的化学去除途径为与OH自由基发生氧化反应;天然源的VOCs如异戊二烯、单萜烯、二甲基硫(DMS)等与NO3的氧化去除贡献十分重要, 在天然源中的贡献甚至超过与OH自由基的氧化反应(Warneke et al., 2004).进一步对比分析了两次观测期间人为源和天然源VOCs在各去除途径的反应速率及其占总去除速率的比例.其中人为源VOCs涵盖了除异戊二烯、单萜烯以外的烃类物质, 天然源VOCs则涵盖了异戊二烯、单萜烯、MVK和MACR等物种.而图 4中大部分OVOCs物种无法直接界定人为源或天然源, 故不纳入两者的计算中.
图 5为城市站点观测期间人为源VOCs及天然源VOCs各去除途径相应去除速率日变化及全天平均贡献比例.可以发现, 人为源和天然源VOCs最重要的去除途径均是OH自由基的氧化去除, 其贡献分别达到了89.4%和81.0%.O3主要与烯烃发生氧化, 分别占人为源和天然源VOCs去除速率的9.5%和8.1%.而NO3因与天然源VOCs反应速率常数较大, 其在天然源去除速率的贡献达到10.9%, 但其在人为源VOCs的贡献仅为1.2%.因此, 城市站点人为源和天然源VOCs去除途径无较明显的差异, 天然源VOCs的NO3的氧化去除途径相对人为源而言会略显重要一些.
 |
| 图 5 城市站点人为源(a)和天然源(b)VOCs物种在大气中被OH、NO3和O3氧化去除的平均去除速率, 人为源(c)和天然源(d)的全天平均的3种途径的去除速率贡献比例 Fig. 5 Average loss rates and 24 h-averaged fractions of loss rates by OH, NO3, O3 oxidation for anthropogenic and biogenic VOCs at the urban site |
区域站点人为源和天然源VOCs各去除途径的反应速率及贡献如图 6所示, 去除速率的贡献比例与城市站点观测有了一定的差异.OH自由基的氧化是人为源VOCs最重要的去除途径, 占比达到了68.7%.由于区域站点NO3自由基浓度更大, 且作为苯乙烯等物质的最主要的去除途径, NO3自由基对人为源VOCs去除速率的贡献更大(26.3%).O3对人为源VOCs去除的贡献在区域站点仅占5.0%.而在天然源VOCs的去除速率贡献比例中, 虽然OH自由基仍以50.6%占据最大的贡献比例, 但NO3自由基氧化的贡献已不容忽视, 占全天总去除速率的37.2%, 其中25.4%是来自于单萜烯的贡献.同时, Geyer等(2001)的研究也表明, 生物源的烃类是NO3自由基最重要的汇.此外, O3也贡献了天然源VOCs化学去除过程的12.2%.因此, 在珠三角区域站点, OH和NO3自由基的氧化在人为源和天然源VOCs的去除均扮演着十分重要的角色.
 |
| 图 6 区域站点人为源(a)和天然源(b)VOCs物种在大气中被OH、NO3和O3氧化去除的平均去除速率, 人为源(c)和天然源(d)的全天平均的3种途径的去除速率贡献比例 Fig. 6 Average loss rates and 24 h-averaged fractions of loss rates by OH, NO3, O3 oxidation for anthropogenic and biogenic hydrocarbons at the regional site |
通过城市站点和区域站点的对比分析, 人为源VOCs在两地最重要的化学去除途径均是与OH自由基的氧化去除, 这与之前Warneke等(2004)的研究相符.但由于区域站点NO3自由基浓度较高, NO3自由基的氧化应得到重视, 这也体现了不同区域VOCs化学去除途径的差异性.对于城市站点而言, 站点周围NO排放源较多;而区域站点附近的近地面NO源较少, 而且从城市传输而来的部分NO也与O3发生滴定反应而去除, 导致区域站点NO浓度较低, 从而为NO3自由基浓度的累积提供了条件.需要注意的是两次观测均在近地面站点开展, 污染物浓度和化学过程受近地面源排放影响较大.最近一些研究表明由于不受近地面排放的影响, 在夜间残留层化学过程与近地面有较大不同, NO3的化学过程和对VOCs的氧化贡献更大一些(Brown et al., 2016; Wang et al., 2018a).
4 结论(Conclusions)1) 烷烃和芳香烃主要是与OH自由基发生反应, 该途径去除贡献最高可达到99%以上;而NO3自由基和O3常与带双键的烯烃发生反应, 贡献特定VOCs物种的去除速率可达80%以上, 特别是一些天然源的烯烃(如单萜烯), 与NO3自由基的氧化是贡献最大的氧化途径.此外, 光解是甲醛最重要的去除途径, 在两个站点均达到了50%以上, 酮类的光解贡献会高于其他OVOCs类物质.
2) 城市和区域站点去除速率的贡献比例对比发现, 烷烃和芳香烃在两地的贡献差距不大, 均为OH自由基的氧化去除途径占主导地位.区域站点的烯烃VOCs与NO3自由基和O3反应的贡献要高于城市站点, 这主要由于在区域站点NO3自由基和O3的浓度均要高于城市站点.特别是NO3自由基在城市站点很大程度上受近地面附近NO排放的影响, 从而也导致了城市站点的NO3自由基浓度相对较低.
3) 对比分析人为源和天然源VOCs各去除途径的贡献发现, 在城市地区OH自由基的氧化在人为源和天然源均为最重要的去除途径(均为80%以上).在区域站点中, 虽然OH自由基氧化仍占据着最大贡献比例, 但NO3自由基氧化的贡献已不容忽视.
Atkinson R. 1997. Gas-phase tropospheric chemistry of volatile organic compounds:1.Alkanes and Alkenes[J]. Journal of Physical and Chemical Reference Data, 26(2): 215-290. |
Atkinson R, Arey J. 2003. Atmospheric Degradation of Volatile Organic Compounds[J]. Chemical Reviews, 103(12): 4605-4638. |
Atkinson R, Aschmann S M, Arey J. 1992. Reactions of hydroxyl and nitrogen trioxide radicals with phenol, cresols, and 2-nitrophenol at (296±2) K[J]. Environmental Science & Technology, 26(7): 1397-1403. |
Atkinson R, Baulch D L, Cox R A, et al. 2006. Evaluated kinetic and photochemical data for atmospheric chemistry:Volume II-gas phase reactions of organic species[J]. Atmospheric Chemistry and Physics, 6(11): 3625-4055. |
Birks J W, Andersen P C, Williford C J, et al. 2018. Folded tubular photometer for atmospheric measurements of NO2 and NO[J]. Atmospheric Measurement Techniques, 11(5): 2821-2835. |
Bo Y, Cai H, Xie S D. 2008. Spatial and temporal variation of emission inventories for historical anthropogenic NMVOCs in China[J]. Atmospheric Chemistry and Physics, 8(3): 11519-11566. |
Brown S S, Dubé W P, Tham Y J, et al. 2016. Nighttime chemistry at a high altitude site above Hong Kong[J]. Journal of Geophysical Research:Atmospheres, 121(5): 2457-2475. |
Brown S S, Stutz J. 2012. Nighttime radical observations and chemistry[J]. Chem Soc Rev, 41(19): 6405-6447. |
Burkholder J, Sander S, Abbatt J, et al. 2015. Chemical kinetics and photochemical data for use in atmospheric studies: evaluation number 18[M]. Pasadena, CA: Jet Propulsion Laboratory, National Aeronautics and Space Administration
|
陈多宏, 钟流举. 2012.珠江三角洲大气超级监测站选址的综合分析与研究[C].中国环境科学学会学术年会.广西南宁: 792-797
|
de Gouw J, Warneke C. 2007. Measurements of volatile organic compounds in the earth's atmosphere using proton-transfer-reaction mass spectrometry[J]. Mass Spectrom Rev, 26(2): 223-257. |
de Gouw J A, Gilman J B, Kim S W, et al. 2017. Chemistry of volatile organic compounds in the los angeles basin:nighttime removal of alkenes and determination of emission ratios[J]. Journal of Geophysical Research:Atmospheres, 122(21): 11843-11861. |
邓雪娇, 王新明, 赵春生, 等. 2010. 珠江三角洲典型过程VOCs的平均浓度与化学反应活性[J]. 中国环境科学, 30(9): 1153-1161. |
Ehhalt D H. 1999. Photooxidation of trace gases in the troposphere Plenary Lecture[J]. Physical chemistry chemical physics, 1(24): 5401-5408. |
Feilberg K L, Johnson M S, Bacak A, et al. 2007. Relative tropospheric photolysis rates of HCHO and HCDO measured at the European photoreactor facility[J]. The Journal of Physical Chemistry, 111(37): 9034-9046. |
Geyer A, Alicke B, Konrad S, et al. 2001. Chemistry and oxidation capacity of the nitrate radical in the continental boundary layer near Berlin[J]. Journal of Geophysical Research, 106(D8): 8013-8025. |
Guenther A, Hewitt C N, Erickson D, et al. 1995. A global model of natural volatile organic compound emissions[J]. Journal of Geophysical Research, 100(D5): 8873-8892. |
Guenther A B, Jiang X, Heald C L, et al. 2012. The model of emissions of gases and aerosols from nature version 2.1 (MEGAN2.1):An extended and updated framework for modeling biogenic emissions[J]. Geoscientific Model Development, 5(6): 1471-1492. |
Guo S, Hu M, Zamora M L, et al. 2014. Elucidating severe urban haze formation in China[J]. Proceedings of the National Academy of Sciences, 111(49): 17373. |
何俊杰, 张国华, 王伯光, 等. 2013. 鹤山灰霾期间大气单颗粒气溶胶特征的初步研究[J]. 环境科学学报, 33(8): 2098-2104. |
Hellén H, Tykkä T, Hakola H. 2012. Importance of monoterpenes and isoprene in urban air in northern Europe[J]. Atmospheric Environment, 59: 59-66. |
Hofzumahaus A, Rohrer F, Lu K, et al. 2009. Amplified trace gas removal in the troposphere[J]. Science, 324(5935): 1702-1704. |
胡珊, 张远航, 魏永杰. 2009. 珠江三角洲大气细颗粒物的致癌风险及源解析[J]. 中国环境科学, 29(11): 1202-1208. |
Jenkin M, Wyche K, Evans C, et al. 2012. Development and chamber evaluation of the MCM v3.2 degradation scheme for β-caryophyllene[J]. Atmospheric Chemistry and Physics, 12(11): 5275-5306. |
Jenkin M E, Saunders S M, Pilling M J. 1997. The tropospheric degradation of volatile organic compounds:a protocol for mechanism development[J]. Atmospheric Environment, 31(1): 81-104. |
Jenkin M E, Young J C, Rickard A R. 2015. The MCM v3.3.1 degradation scheme for isoprene[J]. Atmospheric Chemistry and Physics, 15(20): 11433-11459. |
Kassomenos P A, Dimitriou K, Paschalidou A K. 2013. Human health damage caused by particulate matter PM10 and ozone in urban environments:the case of Athens, Greece[J]. Environmental Monitoring Assessment, 185(8): 6933-6942. |
Keyte I J, Harrison R M, Lammel G. 2013. Chemical reactivity and long-range transport potential of polycyclic aromatic hydrocarbons--a review[J]. Chem Soc Rev, 42(24): 9333-9391. |
Kuang Y, Tao J, Xu W, et al. 2019. Calculating ambient aerosol surface area concentrations using aerosol light scattering enhancement measurements[J]. Atmospheric environment, 216. |
Kuang Y, Zhao C S, Zhao G, et al. 2018. A novel method for calculating ambient aerosol liquid water content based on measurements of a humidified nephelometer system[J]. Atmospheric Measurement Techniques, 11(5): 2967-2982. |
Kumar V, Chandra B P, Sinha V. 2018. Large unexplained suite of chemically reactive compounds present in ambient air due to biomass fires[J]. Sci Rep, 8(1): 626. |
Lau A K, Yuan Z, Yu J Z, et al. 2010. Source apportionment of ambient volatile organic compounds in Hong Kong[J]. Science of the Total Environmental, 408(19): 4138-4149. |
李玲玉, Guenther A B, 顾达萨, 等. 2019. 典型树种挥发性有机物(VOCs)排放成分谱及排放特征[J]. 中国环境科学, 39(12): 4966-4973. |
栗泽苑.2018.华南与华北区域背景大气氧化能力与自由基化学基于观测的模拟研究[D].济南: 山东大学
|
Li Z, Xue L, Yang X, et al. 2018. Oxidizing capacity of the rural atmosphere in Hong Kong, Southern China[J]. Science of the Total Environmental, 612: 1114-1122. |
Ling Z H, Guo H, Cheng H R, et al. 2011. Sources of ambient volatile organic compounds and their contributions to photochemical ozone formation at a site in the Pearl River Delta, southern China[J]. Environmental Pollution, 159(10): 2310-2319. |
Lu K D, Rohrer F, Holland F, et al. 2012. Observation and modelling of OH and HOx concentrations in the Pearl River Delta 2006:a missing OH source in a VOC rich atmosphere[J]. Atmospheric Chemistry and Physics, 12(3): 1541-1569. |
吕子峰, 郝吉明, 段菁春, 等. 2009. 北京市夏季二次有机气溶胶生成潜势的估算[J]. 环境科学, 30(4): 969-975. |
区宇波, 岳玎利, 钟流举, 等. 2013. 中国广东大气超级监测站的规划建设与运行机制研究[J]. 环境科学与管理, 38(11): 63-67. |
Riedel T P, Wolfe G M, Danas K T, et al. 2014. An MCM modeling study of nitryl chloride (ClNO2) impacts on oxidation, ozone production and nitrogen oxide partitioning in polluted continental outflow[J]. Atmospheric Chemistry and Physics, 14(8): 3789-3800. |
Salgado M S, Monedero E, Villanueva F, et al. 2008. Night-Time Atmospheric Fate of Acrolein and Crotonaldehyde[J]. Environmental Science & Technology, 42(7): 2394-2400. |
Sander S, Friedl R, Abbatt J, et al. 2011. Chemical kinetics and photochemical data for use in atmospheric studies, evaluation number 14[J]. JPL publication, 10. |
Saunders S M, Jenkin M E, Derwent R G, et al. 2003. Protocol for the development of the Master Chemical Mechanism, MCM v3 (Part A):tropospheric degradation of non-aromatic volatile organic compounds[J]. Atmospheric Chemistry and Physics, 3(1): 161-180. |
Seinfeld J H, Pandis S N. 2006. Atmospheric chemistry and physics: from air pollution to climate change[M]. Hoboken: John Wiley & Sons, Inc
|
Shao M, Zhang Y, Zeng L, et al. 2009. Ground-level ozone in the Pearl River Delta and the roles of VOC and NOx in its production[J]. Journal of Environmental Management, 90(1): 512-518. |
Solberg S, Dye C, Walker S E, et al. 2001. Long-term measurements and model calculations of formaldehyde at rural European monitoring sites[J]. Atmospheric Environment, 35(2): 195-207. |
唐孝炎, 张远航, 邵敏. 2006. 大气环境化学[M]. .
|
王扶潘, 朱乔, 冯凝, 等. 2014. 深圳大气中VOCs的二次有机气溶胶生成潜势[J]. 中国环境科学, 34(10): 2449-2457. |
Wang H, Lu K, Chen X, et al. 2017. High N2O5 Concentrations Observed in Urban Beijing:Implications of a Large Nitrate Formation Pathway[J]. Environmental Science & Technology Letters, 4(10): 416-420. |
Wang H, Lu K, Chen X, et al. 2018a. Fast particulate nitrate formation via N2O5 uptake aloft in winter in Beijing[J]. Atmospheric Chemistry and Physics, 18(14): 10483-10495. |
Wang H, Lu K, Guo S, et al. 2018b. Efficient N2O5 uptake and NO3 oxidation in the outflow of urban Beijing[J]. Atmospheric Chemistry and Physics, 18(13): 9705-9721. |
Warneke C, de Gouw J A, Goldan P D, et al. 2004. Comparison of daytime and nighttime oxidation of biogenic and anthropogenic VOCs along the New England coast in summer during New England Air Quality Study 2002[J]. Journal of Geophysical Research, 109(D10309). |
Wei W, Wang S, Chatani S, et al. 2008. Emission and speciation of non-methane volatile organic compounds from anthropogenic sources in China[J]. Atmospheric Environment, 42(20): 4976-4988. |
Williams J. 2004. Organic trace gases in the atmosphere:An overview[J]. Environmental Chemistry, 1(3): 125-136. |
Wolfe G M, Marvin M R, Roberts S J, et al. 2016. The Framework for 0-D Atmospheric Modeling (F0AM) v3.1[J]. Geoscientific Model Development, 9(9): 3309-3319. |
Wu F, Yu Y, Sun J, et al. 2016. Characteristics, source apportionment and reactivity of ambient volatile organic compounds at Dinghu Mountain in Guangdong Province, China[J]. Science of the Total Environmental, 548-549: 347-359. |
尹鸿鸣, Kable S H. 2008. 大气污染物甲醛光解离动力学的研究进展[J]. 科学通报, 53(1): 69-73. |
Yuan B, Hu W W, Shao M, et al. 2013. VOC emissions, evolutions and contributions to SOA formation at a receptor site in eastern China[J]. Atmospheric Chemistry and Physics, 13(17): 8815-8832. |
Yuan B, Koss A R, Warneke C, et al. 2017. Proton-Transfer-Reaction Mass Spectrometry:Applications in Atmospheric Sciences[J]. Chemical Reviews, 117(21): 13187-13229. |
郑君瑜, 袁斌, 王俏巧, 等. 2019. 区域空气质量改善的历史经验和挑战——代"第五届经济快速发展地区空气质量改善国际学术研讨会"专刊序[J]. 环境科学学报, 39(1): 3-5. |