 2019, Vol. 39
2019, Vol. 39



2. 云南省土壤固碳与污染控制重点实验室, 昆明 650500
2. Yunnan Provincial Key Laboratory of Carbon Sequestration and Pollution Control in Soils, Kunming 650500
光催化降解水中的污染物已经被广泛研究并应用, 其中, 有关光催化剂的催化机理也形成了共识性的理解(Hsieh et al., 2010; Mohapatra et al., 2014; Wang et al., 2018).其核心机制为半导体光催化材料受到光激发产生光生电子(e-)和空穴(h+), 具备氧化还原性质的e-及h+进一步利用溶液中的水分子或氧气生成氧化还原活性物质(Reactive Oxygen Species, ROS), 如羟基自由基(·OH)和超氧阴离子自由基(·O2-), 这类氧化能力强的自由基便是降解污染物的重要反应活性物质(Horikoshi et al., 2003; Janczyk et al., 2006; Kim et al., 2010).因此, 提高污染物的降解效率, 其实质在于增加ROS与污染物的反应机会.目前研究证实了不同种类ROS会选择性地降解某些污染物, 甚至同一种自由基, 如TiO2产生的·OH也会表现出对污染物的选择性降解(Minero et al., 2000; Lv et al., 2016).这种选择性降解主要基于其在催化剂表面的吸附选择性, 因为光催化剂表面产生的ROS会优先与吸附在其表面的污染物发生反应(Park et al., 2004a; 2004b; Lv et al., 2006; Sheng et al., 2013).因此, 如何提高弱吸附污染物的降解及增强ROS利用效率有待深入研究.
TiO2作为一种研究最多、应用最广的光催化材料, 因其禁带宽度(3.2 eV)可满足转化水分子生成·OH的电势能而被广泛运用于到污染物的降解中(石建稳等, 2014; 刘文婧等, 2016).Park等(2004b)研究发现, TiO2光激发产生的自由基可以穿透高分子膜并降解膜另一面的罗丹明B染料.该研究提出了一个新的思考:光催化剂不仅能降解表面吸附的物质, 自由基还可以向外迁移并与没有吸附在催化剂颗粒表面的物质反应.之后有研究者推断出·OH在TiO2表面的形成机制, 并认为·OH可划分为表面束缚态·OH和游离态·OH (free ·OH) (Nosaka et al., 2003; Murakami et al., 2007; Yu et al, 2009; Liao et al., 2013; Biljana et al., 2015).Kondrakov等(2016)还利用同位素示踪的方法, 证实了游离·OH对降解的贡献.因此本文提出假设, 如果能有效提高游离自由基的产量, 弱吸附有机物的降解效率可以被相应提高.
为了探究通过增加游离自由基提高弱吸附有机物降解的机制, 本文系统设计了TiO2光催化降解弱吸附间苯二酚(RE)的实验, 以制备的高暴露{001}晶面的TiO2作为模型晶面结构, 利用氟离子对该晶面的强吸附作用竞争表面羟基位点, 从而提高游离自由基的生成.同时, 通过一系列ROS的猝灭降解实验, 以及EPR检测·OH和·O2-的产量, 全面讨论氟离子作用下自由基种类和数量的变化, 明确游离·OH对弱吸附RE降解的关键作用, 以期为弱吸附有机污染物的降解提供思路和科学依据.
2 实验方法(Experimental methods) 2.1 材料与表征钛酸丁酯、乙醇、氢氧化钠、间苯二酚购于美国Sigma公司;氢氟酸(HF)、氟化钠(NaF)、甲醇(CH3OH)、苯醌(BQ)、5, 5-二甲基-1-吡咯啉-N-氧化物(DMPO)、磷酸盐缓冲液(PBS)购于上海阿拉丁生化科技股份有限公司.所有试剂均为化学纯或分析纯, 无需进一步纯化或处理.所有实验用水均为超纯水.
高暴露{001}晶面TiO2样品({001}-TiO2)通过水热法合成(Li et al., 2015).合成方法如下:25 mL钛酸丁酯与3 mL氢氟酸在干燥的100 mL聚四氟乙烯反应釜内衬中均匀混合, 放入高压反应釜并转移至烘箱中200 ℃保持24 h, 所得白色产物用超纯水和乙醇交替洗涤至pH接近中性;将粉末浸泡于100 mL NaOH(0.1 mmol·L-1)溶液中过夜, 再次用超纯水及乙醇交替洗涤5次, 最终产物离心并干燥.通过X射线衍射(XRD, D/max-2200 diffractometer)、透射电子显微镜(TEM/HRTEM, F30 S-TWIN TECNAI G2, FEI)和比表面仪(Micromeritics ASAP 2020)对{001}-TiO2样品进行晶体结构和形貌尺寸表征分析.
2.2 光催化降解实验以{001}-TiO2光催化降解间苯二酚(RE)为例, 称取10 mg {001}-TiO2粉末均匀分散于50 mL预先配置好的RE水溶液中(RE浓度分别为0.454、0.091 mmol·L-1), 调节pH至4.5, 在避光条件下磁力搅拌1 h达到吸附平衡.光催化降解实验在带有磁力搅拌的旋转光化学反应器中进行, 光源为配备循环冷却水装置的300 W汞灯(上海亚明), 辐射强度3750 μW·cm-2(距离7.5 cm).光照时间间隔0、5、15、30、50、70、90、120 min时取样, 过0.45 μm滤膜除去固体颗粒, 使用高效液相色谱(HPLC, Agilent Technologies 1260 Infinity)分析RE浓度.猝灭降解实验的方法步骤同上, 仅在混合RE溶液的同时加入NaF(5 mmol·L-1)、甲醇(0.074 mmol·L-1)、苯醌(0.074 mmol·L-1), 调节pH至4.5.空白对照实验为相同条件下RE在纯水中的光降解.
2.3 F-吸附实验称取1.6 mg {001}-TiO2与8 mL浓度梯度为0.5、1、2、2.5、3、4、5、6 mmol·L-1的NaF溶液混合, 超声分散5 min后置于避光环境中搅拌24 h.吸附平衡后, 取样并通过0.45 μm滤膜除去催化剂颗粒, 经离子色谱(IC, 岛津LC20AD)检测溶液中的F-浓度, 即平衡浓度Ce.初始F-浓度C0与平衡浓度Ce的差值与{001}-TiO2质量比为吸附量Qe.实验结果符合Langmuir吸附方程:
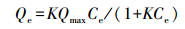
|
(1) |
式中, Qmax为最大吸附量(mmol·g-1), K为平衡常数(mmol-1).
2.4 EPR检测·OH和·O2-采用电子顺磁共振光谱仪(EPR, Bruker, A300-6/1)捕获并检测·OH和·O2-.在光催化反应过程中用注射器取100 μL TiO2悬浮液与100 μL 0.3 mmol·L-1 DMPO溶液(由0.01 mmol·L-1 PBS配置)混合, 注入石英细管中光照并检测·OH信号.采用等体积比的甲醇和0.01 mmol·L-1 PBS配置的DMPO混合溶液作为·O2-捕获剂(Yao et al, 2013).EPR测试参数为:调制频率1.00 kHz, 调制振幅1.00 G, 微波频率9.2~9.9 GHz, 功率为9 dB(或21 mW), 扫描时间为20.48 ms.
3 结果与讨论(Results and discussion) 3.1 {001}晶面TiO2样品的形貌及结构表征为了构建均匀的晶体表面结构, 本文采用水热法制备了高暴露{001}晶面的TiO2样品, 该晶面已知的原子结构可作为探究有机分子吸附的模型晶面(Li et al., 2015), 同时也可提供优良的光催化活性(Tian et al., 2012; Ye et al., 2013).图 1a为{001}-TiO2粉末样品的X射线衍射图谱.如图所示, 样品所有衍射峰位置和强度都与锐钛型TiO2(JCPDS card no. 21-1272)的特征衍射峰一致.同时, XRD图谱中没有其他衍射峰出现, 说明样品中无其他晶体生成, 表明所得样品为纯相锐钛矿TiO2.其中, 主要特征衍射峰, 如(101)、(200)的峰形窄且峰强高, 说明所得晶体结晶度优良.图 1b为氮气吸附脱附法测试{001}-TiO2样品的比表面积和孔径分布.通过BET法测得样品的比表面积为153.52 m2·g-1, 较高的比表面积说明样品粒径应该为纳米级.如图 1b插图所示, 采用Barrett-Joyner-Halenda法计算得出样品的孔径分布为10~30 nm, 这是由样品晶片堆叠而形成的空隙结构.
 |
| 图 1 {001}-TiO2粉末样品的XRD图谱(a)和N2吸附脱附等温线及孔径分布曲线(插图)(b) Fig. 1 XRD pattern of {001}-TiO2 powder(a) and nitrogen adsorption-desorption isotherms and the corresponding pore size distribution curve (inset) of {001}-TiO2 powder(b) |
进一步通过TEM和HRTEM观察{001}晶面TiO2的形貌和结构.如图 2a所示, 样品均为规则的四方片状.经测量和统计, 四面体片的边长和厚度分别约为81 nm和5 nm, 说明最大暴露面占晶体总面积的89%以上.在单个晶片的HRTEM图中可以观察到同一方向平行排列的晶格条纹, 其条纹间距0.235 nm与XRD对应的标准卡片(JCPDS card no. 21-1272)中(001)晶面d值相吻合, 直接证实了{001}-TiO2样品为高比例暴露{001}晶面的TiO2晶片.
 |
| 图 2 {001}-TiO2样品的TEM(a)和HRTEM(b) Fig. 2 TEM (a) and HRTEM(b) images of {001}-TiO2 sample |
苯酚类物质的光催化降解在以往的研究中被广泛报道, 其降解中间产物主要为邻苯二酚、对苯二酚及间苯二酚(Lv et al., 2016).值得注意的是, 这3种羟基加成的中间产物吸附性能发生了巨大变化, 邻苯二酚可以与TiO2吸附及络合(Gulley-Stahl et al., 2010), 因此, 能够进一步被高效地光催化降解;而间苯二酚与TiO2的吸附变弱, 可能致使进一步降解延缓并难以彻底矿化.因此, 本研究首先进行了{001}-TiO2光催化降解RE和RE自身光降解的对比实验, 其浓度变化如图 3所示.在光催化体系RE+TiO2中, 无论RE初始浓度高(0.454 mmol·L-1)或低(0.091 mmol·L-1), RE的降解效果没有明显提高, 甚至出现抑制的现象.该现象源于两方面原因:①{001}-TiO2催化产生的反应活性物种无法接触到弱吸附的间苯二酚;②{001}-TiO2对紫外光产生了屏蔽和散射效应从而降低了RE自身的光降解速率.
 |
| 图 3 RE与RE+TiO2光催化体系的降解效果比较 Fig. 3 The degradation performance comparison between RE and RE+TiO2 photocatalytic system |
为了提高间苯二酚的光催化降解效率, 本研究拟在光催化降解体系中加入NaF.因为F-能够竞争TiO2表面羟基的吸附位点(Xu et al., 2007; Montoya et al., 2010), 促使更多的·OH从TiO2表面脱落并游离到溶液中.F-在25 ℃、pH=4.5条件下与{001}-TiO2反应得到的吸附等温线如图 4a所示, 吸附曲线符合Langmuir吸附方程(式(1)), 计算结果见表 1.F-在{001}-TiO2上的饱和吸附量达到2.782 mmol·g-1, 该数值远高于商业P25 TiO2上F-的饱和吸附量(Muggli et al., 2001), 这主要归功于高暴露{001}晶面为F-提供了大量的结合位点.
 |
| 图 4 F-在{001}-TiO2表面的吸附等温线(a)和F-对RE在{001}-TiO2上吸附的影响(b) Fig. 4 Adsorption isotherms of F- on {001}-TiO2 (a) and effect of F- on the adsorption of RE on {001}-TiO2(b) |
| 表 1 Langmuir方程计算结果 Table 1 Calculation results by Langmuir equation |
图 4b为25 ℃、pH=4.5条件下RE在{001}-TiO2上的吸附等温线, 并与加入NaF后的吸附等温线进行比较.随着平衡浓度(Ce)的增加, {001}-TiO2上RE的吸附量(Qe)增大并达到平衡, 吸附曲线同样符合Langmuir吸附方程式(1).计算结果显示(表 1), RE的最大吸附量仅0.015 mmol·g-1, 加入F-时, RE在{001}-TiO2表面的最大吸附量仅降低0.003 mmol·g-1, 说明大量吸附在{001}-TiO2表面的F-对RE吸附的影响很微弱.因此, 针对在{001}-TiO2上吸附很弱的RE, F-的添加并能不改变RE在体系中的吸附性质.
3.3 {001}-TiO2光催化降解间苯二酚过程中的反应活性物质以往关于TiO2光催化机理的研究已经明确其较高的污染物降解能力来自于光激发后进一步产生的ROS, 如·OH、·O2-、h+(Kuang et al., 2016).为了明确该光催化体系中的ROS种类和产生路径, 本文设置了一系列自由基猝灭实验, 在反应体系中加入甲醇(CH3OH)和苯醌(BQ)可以分别猝灭·OH和·O2-.如图 5所示, 在加入甲醇的体系中, RE的降解被明显抑制, 说明猝灭溶液中的·OH能显著影响RE的降解, 而·O2-的猝灭(加入苯醌)对RE降解影响微弱.因此, {001}-TiO2光催化降解RE体系中, ·OH是主要的反应活性物质.
 |
| 图 5 不同猝灭剂条件下RE的光催化降解性能比较 Fig. 5 Comparison of RE photodegradation performance in the presence of different scavengers |
同时, 本研究通过EPR检测相同条件下光催化反应体系中·OH和·O2-的产生情况.如图 6所示, 不加入光催化剂{001}-TiO2时, 纯RE溶液在紫外光照射下没有自由基信号产生, 说明RE自身光降解不会引发自由基生成.在{001}-TiO2光催化降解RE的体系中, EPR检测到强度比为1:2:2:1的四重峰, 该特征峰为DMPO捕获·OH形成的DMPO\OH加合物信号.采用·O2-捕获方法通过EPR测得峰强比为1:1:1:1:1:1的六重峰信号, 该六重峰为·O2-的特征峰.以上实验结果证实了{001}-TiO2光催化降解RE体系中同时存在·OH和·O2-, RE的降解主要取决于·OH.
 |
| 图 6 ·OH和·O2-的特征EPR信号 Fig. 6 Characteristic EPR signals of ·OH and ·O2- |
由前述实验可知, {001}-TiO2光催化降解RE的效果很差, 降解速率甚至低于RE的自身光降解, 因此, 本研究采用F-与{001}-TiO2表面形成的自由基发生竞争吸附(Lv et al., 2012), 试图促使自由基游离到溶液中增加与RE的反应机会.图 7显示, 在{001}-TiO2光催化体系中加入5 mmol·L-1 NaF能极大地提高RE的光催化降解效率.在光照30 min时, RE-TiO2-F-体系的降解率达到了94%, 远高于纯RE(64%)和RE-TiO2体系(50%).当RE降解率达到95%时, F-的加入使RE-TiO2光催化体系的降解速率提高了3倍.
 |
| 图 7 F-对光催化降解RE的促进作用 Fig. 7 The enhanced photodegradation of RE by F- |
为了证实F-促使RE降解率提高是基于溶液相中自由基的增加, 本研究设计了更细致的自由基猝灭实验, 即在F-参与的{001}-TiO2光催化降解体系中使用甲醇、苯醌(BQ)分别猝灭体系中的·OH和·O2-.如图 8所示, 相比在120 min内RE完全降解的TiO2-F-体系, ·OH和·O2-猝灭剂的加入都不同程度地抑制了RE的降解.当TiO2-F-光催化体系中猝灭了·O2-, 紫外光照120 min后RE的降解量有所下降, 但仍能达到70.4%, 说明在TiO2-F-光催化体系中·O2-对RE的降解有微弱贡献.当TiO2-F-体系中添加了·OH猝灭剂(CH3OH)之后, RE的光催化降解被显著抑制, 说明·OH是降解RE最主要的活性物质.值得注意的是, F-相比表面OH或·OH在{001}-TiO2上有更强的吸附性(Lv et al., 2006), F-占据{001}-TiO2表面吸附位点后, 使表面产生的·OH无法竞争吸附在催化剂表面, 因此, F-也可视为表面·OH的去除剂.如图 8所示, TiO2-F--CH3OH体系中催化剂表面及溶液中游离的·OH被同时猝灭, RE的降解几乎完全被抑制.与之相比, 在仅猝灭游离·OH的TiO2-CH3OH体系中, RE的降解抑制程度非常接近, 进一步证实了溶液中游离的·OH是降解RE的主要活性物质.
 |
| 图 8 TiO2-F-光催化体系中不同猝灭剂条件下RE的降解性能比较 Fig. 8 Photodegradation performance of RE by TiO2-F- system in the presence of different scavengers |
本研究还采用EPR对光催化反应进程中特定时间点的·OH及·O2-捕获量进行检测, 自由基强度值选取·OH特征峰的第2个峰和·O2-特征峰的第1个峰, 峰强度可以反映同一种自由基的浓度大小.如图 9a和9c所示, 经4 min的自由基连续监测发现, F-的加入均能显著提高·OH和·O2-的浓度, 再次验证了F-提高RE降解速率的机理为:F-与{001}-TiO2表面羟基及·OH的竞争吸附促使游离·OH增加, 进而使弱吸附性的RE可以在溶液中被有效地降解.同时, 溶液中·O2-的浓度也在F-加入条件下增加, 这是因为F-与表面Ti原子的强吸附使{001}-TiO2表面呈负电, 使迁移到表面的光生e-更多地保留下来与溶液中的O2反应生成·O2- (如反应式(2)~(6)所示).因此, 通过EPR自由基检测实验发现, F-不仅能提高·OH产量, 也能提高·O2-产量, 并且其促进产生的·OH主要游离在溶液中, 更利于弱吸附有机污染物的降解反应.
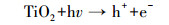
|
(2) |
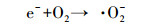
|
(3) |
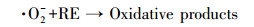
|
(4) |

|
(5) |
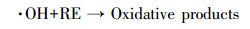
|
(6) |
 |
| 图 9 加入F-条件下·OH(a、b)和·O2-(c、d)的EPR信号强度比较 Fig. 9 EPR signal intensities for ·OH (a, b) and ·O2-(c, d) in the presence of F- |
1) RE在{001}-TiO2表面的吸附能力很弱, 并且光催化降解效果相比RE自身的光降解没有明显提高, 说明弱吸附的RE与{001}-TiO2表面生成的自由基反应效率很低.
2) 在光催化体系中加入F-, F-在强亲水的{001}-TiO2表面吸附量很大, 同时对RE的吸附量影响微弱, 但RE的光催化降解效率却提升了近3倍.通过自由基猝灭降解实验发现, ·OH和·O2-为该体系的氧化活性物质, RE的降解主要取决于游离在溶液中的·OH浓度.其机理为:F-的强吸附作用竞争了光催化产生的·OH在{001}-TiO2表面的吸附位点, 使更多的·OH脱离颗粒表面游离到溶液中并与RE反应.
3) EPR对光催化反应过程中·OH和·O2-产量的检测结果表明, F-不仅可以促进游离·OH的产生, 还能激发产生更多的·O2-.·O2-来源于有效分离的光生e-与O2或·OH的反应.
综上, 提高溶液中的游离·OH产量能明显提升弱吸附有机物的光催化降解效率, 该思路可广泛运用到光催化或芬顿法处理污水等领域.
Biljana A, Vesna D, Daniela Š, et al. 2015. Mechanism of clomazone photocatalytic degradation:hydroxyl radical, electron and hole scavengers[J]. Reaction Kinetics, Mechanisms and Catalysis, 115(1): 67–79.
DOI:10.1007/s11144-014-0814-z
|
Gulley-Stahl H, Hogan P A, Schmidt W L, et al. 2010. Surface complexation of catechol to metal oxides:an ATR-FTIR, adsorption, and dissolution study[J]. Environmental Science & Technology, 44: 4116–4121.
|
Horikoshi S, Hidaka H, Serpone N. 2003. Hydroxyl radicals in microwave photocatalysis.Enhanced formation of OH radicals probed by ESR techniques in microwave-assisted photocatalysis in aqueous TiO2 dispersions[J]. Chemical Physics Letters, 376: 475–480.
DOI:10.1016/S0009-2614(03)01007-8
|
Hsieh W P, Pan J R, Huang C, et al. 2010. Enhance the photocatalytic activity for the degradation of organic contaminants in water by incorporating TiO2 with zero-valent iron[J]. Science of the Total Environment, 408: 672–679.
DOI:10.1016/j.scitotenv.2009.07.038
|
Janczyk A, Krakowska E, Stochel G, et al. 2006. Singlet oxygen photogeneration at surface modified titanium dioxide[J]. Journal of the American Chemical Society, 128: 15574–15575.
DOI:10.1021/ja065970m
|
Kim J, Choi W, Park H. 2010. Effects of TiO2 surface fluorination on photocatalytic degradation of methylene blue and humic acid[J]. Research on Chemical Intermediates, 36: 127–140.
DOI:10.1007/s11164-010-0123-8
|
Kondrakov A, Ignatev A N, Lunin V V, et al. 2016. Roles of water and dissolved oxygen in photocatalytic generation of free OH radicals in aqueous TiO2 suspensions:An isotope labeling study[J]. Applied Catalysis B:Environmental, 182: 424–430.
DOI:10.1016/j.apcatb.2015.09.038
|
Kuang L Y, Zhao Y P, Zhang W, et al. 2016. Roles of reactive oxygen species and holes in the photodegradation of cationic and anionic dyes by TiO2 under UV Irradiation[J]. Journal of Environmental Engineering, 142(2): 04015065.
DOI:10.1061/(ASCE)EE.1943-7870.0001032
|
Li C H, Koenigsmann C, Ding W D, et al. 2015. Facet-dependent photoelectrochemical performance of TiO2 nanostructures:an experimental and computational study[J]. Journal of the American Chemical Society, 137: 1520–1529.
DOI:10.1021/ja5111078
|
Liao H D, Reitberger T. 2013. Generation of free OHaq radicals by black light illumination of degussa(evonik) P25 TiO2 aqueous suspensions[J]. Catalysts, 3: 418–443.
DOI:10.3390/catal3020418
|
刘文婧, 景传勇. 2016. 纳米二氧化钛光催化转化甲基砷的研究[J]. 环境科学学报, 2016, 36(1): 172–177.
|
Lv K L, Cheng B, Yu J G, et al. 2012. Fluorine ions-mediated morphology control of anatase TiO2 with enhanced photocatalytic activity[J]. Physical Chemistry Chemical Physics, 14: 5349.
DOI:10.1039/c2cp23461k
|
Lv K L, Guo X J, Wu X F, et al. 2016. Photocatalytic selective oxidation of phenol to produce dihydroxybenzenes in a TiO2/UV system:hydroxyl radical versus hole[J]. Applied Catalysis B:Environmental, 199: 405–411.
DOI:10.1016/j.apcatb.2016.06.049
|
Lv K L, Xu Y M. 2006. Effects of polyoxometalate and fluoride on adsorption and photocatalytic degradation of organic dye X3B on TiO2:the difference in the production of reactive species[J]. Journal of Physical Chemistry B, 110: 6204–6212.
DOI:10.1021/jp055228t
|
Minero C, Mariella G, Maurino V, et al.2000.Photocatalytic transformation of organic compounds in the presence of inorganic ions.2.Competitive reactions of phenol and alcohols an a titanium dioxide-fluoride system[J].Langmuir, 16: 8964-8972
https://pubs.acs.org/doi/abs/10.1021/la0005863 |
Mohapatra D P, Brar S K, Daghrir R, et al. 2014. Photocatalytic degradation of carbamazepine in wastewater by using a new class of whey-stabilized nanocrystalline TiO2 and ZnO[J]. Science of the Total Environment, 485: 263–269.
|
Montoya J F, Salvador P. 2010. The influence of surface fluorination in the photocatalytic behaviour of TiO2 aqueous dispersions:An analysis in the light of the direct-indirect kinetic model[J]. Applied Catalysis B:Environmental, 94: 97–107.
DOI:10.1016/j.apcatb.2009.10.025
|
Muggli D S, Ding L F. 2001. Photocatalytic performance of sulfated TiO2 and Degussa P-25 TiO2 during oxidation of organics[J]. Applied Catalysis B-Environmental, 32: 181–194.
DOI:10.1016/S0926-3373(01)00137-0
|
Murakami Y, Endo K, Ohta I, et al. 2007. Can OH radicals diffuse from the UV-Irradiated photocatalytic TiO2 surfaces? laser-induced-fluorescence study[J]. Journal of Physical Chemistry C, 111: 11339–11346.
DOI:10.1021/jp0722049
|
Nosaka Y, Komori S, Yawata K, et al. 2003. Photocatalytic ·OH radical formation in TiO2 aqueous suspension studied by several detection methods[J]. Physical Chemistry Chemical Physics, 5(20): 4731–4735.
DOI:10.1039/B307433A
|
Park H, Choi W. 2004a. Effects of TiO2 surface fluorination on photocatalytic reactions and photoelectrochemical behaviors[J]. Journal of Physical Chemistry B, 108: 4086–4093.
DOI:10.1021/jp036735i
|
Park J S, Choi W. 2004b. Remote photocatalytic oxidation mediated by active oxygen species penetrating and diffusing through polymer membrane over surface fluorinated TiO2[J]. Chemistry Letters, 34(12): 1630–1631.
|
Sheng H, Li Q, Ma W H, et al. 2013. Photocatalytic degradation of organic pollutants on surface anionized TiO2:Common effect of anions for high hole-availability by water[J]. Applied Catalysis B:Environmental, 138-139: 212–218.
DOI:10.1016/j.apcatb.2013.03.001
|
石建稳, 艾慧颖, 王旭, 等. 2014. TiO2/粉煤灰光催化降解双氯芬酸钠研究[J]. 环境科学学报, 2014, 34(2): 370–376.
|
Tian F, Zhang Y P, Zhang J, et al. 2012. Raman spectroscopy:a new approach to measure the percentage of anatase TiO2 exposed(001) facets[J]. Journal of Physical Chemistry C, 116: 7515–7519.
DOI:10.1021/jp301256h
|
Wang H M, You C F, Tan Z C. 2018. Enhanced photocatalytic oxidation of SO2 on TiO2 surface by Na2CO3 modification[J]. Chemical Engineering Journal, 350: 89–99.
DOI:10.1016/j.cej.2018.05.128
|
Xu Y M, Lv K L, Xiong Z G, et al. 2007. Rate enhancement and rate inhibition of phenol degradation over irradiated anatase and rutile TiO2 on the addition of NaF:new insight into the mechanism[J]. Journal of Physical Chemistry C, 111: 19024–19032.
DOI:10.1021/jp076364w
|
Yao Y Y, Wang L, Sun L J, et al. 2013. Efficient removal of dyes using heterogeneous Fenton catalysts based on activated carbon fibers with enhanced activity[J]. Chemical Engineering Science, 101: 424–431.
DOI:10.1016/j.ces.2013.06.009
|
Ye H P, Lu S M. 2013. Photocatalytic selective oxidation of phenol in suspensions of titanium dioxide with exposed {001} facets[J]. Applied Surface Science, 277: 94–99.
DOI:10.1016/j.apsusc.2013.04.008
|
Yu J G, Wang W G, Cheng B, et al. 2009. Enhancement of photocatalytic activity of mesporous TiO2 powders by hydrothermal surface fluorination treatment[J]. Journal of Physical Chemistry C, 113(16): 6743–6750.
DOI:10.1021/jp900136q
|