 2018, Vol. 38
2018, Vol. 38



离子液体(ILs)是由不对称的有机阳离子与有机或无机阴离子构成的一类新型溶剂(Gutiérrez et al., 2018).与传统的有机溶剂相比, 具有热稳定性好、导电性好、低熔点、低蒸汽压和电化学窗口宽等优良性能.目前, 离子液体已广泛应用于有机催化、生物催化、聚合作用、萃取等领域(Dreyfus et al., 1976; Ventura et al., 2009; Khan et al., 2017; Vekariya, 2017; Costa et al., 2017).尽管离子液体拥有许多优点, 常被称为“绿色溶剂”, 但离子液体本身并非完全无毒性(Dyke et al., 1972; 张瑾等, 2013).离子液体由于无挥发性, 因此不会对大气造成污染, 但大部分离子液体具有较高的水溶性及低生物降解性, 随着离子液体的流失和废弃就会对生态系统造成潜在危害(Wang et al., 2017).咪唑类离子液体在基础研究和工业化生产中的应用最为广泛, Cho等(2012)报道了不同阴离子种类的离子液体的藻毒性顺序, 表明在卤素原子中溴离子的藻毒性大于其他阴离子.
大量研究报道表明咪唑类离子液体对微生物、植物、水生生物等具有不同程度的毒性(井长勤等, 2013; Liu et al., 2015; Liu et al., 2017). Nancharaiah和Francis(2011)评估了咪唑类离子液体对梭状芽孢杆菌属生长的抑制作用, 发现[C2mim]OAc对革兰阳氏梭菌表现出促进作用, 即离子液体浓度低于2.5 g· L-1时促进革兰阳氏梭菌的生长和发酵, 而高于此浓度则表现出抑制作用. Du等(2012)发现将斑马鱼暴露于1-辛基-3-甲基咪唑六氟硼酸盐[C8mim]PF6中, 其体内的活性氧基团含量发生变化, 尤其是两个关键的抗氧化酶(氧化还原酶和超氧化物歧化酶)的活性在高浓度(40 g·L-1)离子液体中显著降低, 此外, 在离子液体中斑马鱼的DNA和脂质体同样受到损伤.
关于离子液体的生物毒性、环境毒性已经有大量的研究报道, 但离子液体的基因毒性研究相对较少. Chandran等(2012)利用分子动力学(Molecular dynamics, MD)模拟、圆二色谱和荧光染色法发现离子液体侵入DNA双股螺旋小沟, 扰乱了DNA周围的水笼, 抑制DNA的水解而使其长期保持本身构型. Ding等(2010)系统地研究了[C4mim]Cl与DNA相互作用的结合特性和分子机制. 31P NMR及FT-IR的结果表明, 阳离子与DNA磷酸基团之间存在静电吸引, 能使[C4min]Cl有效地结合到DNA中形成DNA-ILs复合物; [C4mim]+中的烷基链与DNA碱基对间发生疏水结合, [C4mim]+附于DNA表面.此外, 当离子液体浓度低于0.06 mol· L-1时, [C4mim]+的头部基团通过静电作用伸入到DNA的磷酸基团内, 而烷基侧链通过疏水作用平行地排列在DNA表面; 当离子液体浓度大于0.06 mol· L-1时, 阳离子烷基侧链则垂直地附于DNA表面. Jumbri等(2014)通过MD模拟和实验发现在纯的[C2mim]Br、[C4mim]Br、[C6mim]Br离子液体中, DNA头部和尾部的碱基对受离子液体影响较大, 而DNA双螺旋结构中的磷酸基团受离子液体的影响较小.
核酸碱基是DNA和RNA分子的重要组成部分, 在基因遗传信息传递方面起着主导作用, 是引起生物遗传变异的基础性物质.胞嘧啶作为DNA和RNA分子共同拥有的嘧啶类碱基, 得到广泛的关注与应用.胞嘧啶存在多种互变异构体, 但它们在核酸分子中主要以最稳定的异构体形式存在. Ueda等(1963)、Dreyfus等(1976)和Tomic等(2005)发现胞嘧啶在水溶液中以及以固相存在时, 氨基-羰基-N1H异构体是其主要异构体.
密度泛函理论(Density functional theory, DFT)已被广泛地应用于有机物的计算.吴阳等(2016)采用DFT方法和Born-Fajans-Haber循环法对咪唑醋酸离子液体的热力学性质进行详细计算, 结果与实验值具有较好的一致性.本文通过DFT理论计算阳离子(C2/4mim+)、阴离子(Br-)和溴化咪唑类离子液体([C2/4mim]Br)对胞嘧啶(C)结构和性质影响及其两者之间的结合特性, 有助于深入了解两者之间的作用本质. 图 1是本文研究体系的原子序号标识图和DFT计算的静电势图, 为后文的分析和作用结构设计基础.
 |
| 图 1 胞嘧啶和咪唑阳离子的结构示意图和静电势图 Fig. 1 Schematic structures of cytosine and cations, the corresponding electrostatic potential surfaces for them are presented |
本文采用M06-2X和ωB97XD方法在6-311++G(2d, p)基组水平, 对[C-C2/4mim+]、[C-Br-]和[C-[C2/4mim]Br]体系的几何结构、能量学特征和谱学等性质进行计算.文献表明M06-2X和ωB97XD方法在计算弱相互作用时, 其计算结果与实验值吻合较好(Chai et al., 2008; Chai et al., 2009; Grimme, 2011).在优化结构的基础上, 利用分子中原子理论(AIM) (Bader, 1990)探讨离子液体与胞嘧啶之间相互作用的电子密度拓扑信息, 同时采用自然键轨道分析(NBO)(Vasiliy et al, 2004.)胞嘧啶和离子液体的轨道对作用体系中稳定化能的贡献, 并采用极化连续介质模型(PCM)(Orozco et al., 2000)研究水溶液对3种作用体系的影响.在计算复合物相互作用能时, 考虑了零点振动能(Zero-point vibrational energy, ZPVE)和基组重叠误差(Basis set superposition errors, BSSE)校正, BSSE校正采用Counterpoise(CP)方法.所有计算和分析都是用G 09程序包(Frisch et al., 2013)和Multiwfn软件完成(Lu et al., 2012).
2.1 几何结构和能量学特征 2.1.1 [C-C2/4mim+]作用体系基于图 1中静电势图,设计初始结构在M06-2X/6-311++G(2d, p)和ωB97XD/6-311++G(2d, p)水平对胞嘧啶和C2mim+、C4mim+作用体系进行优化, 各自得到12种稳定结构.各结构分别以M06-2X/6-311++G(2d, p)水平计算结果为基准, 按其电子相对能量由低到高依次编号C-C2/4mim-m (m=1~12), 胞嘧啶与两种阳离子相互作用最稳定结构及主要原子间的距离都在图 2中标明从上往下依次为M06-2X和ωB97XD方法在气相和水相中的计算结果, 胞嘧啶单体及其在最稳定复合物中结构参数进行了对比.下文分析括号内的数值为ωB97XD方法的计算结果.两种水平下计算的能量学数据见表 1, 电子总能量ET是考虑了零点振动能(ZPVE)校正的数值, 其中相对能(ER)、结合能(EB/EBSSE-B)、变形能(ED)分别由下式给出:
 |
| 图 2 最稳定的C-C2mim-1和C-C4mim-1的几何结构 Fig. 2 Geometrical structures of the most stable C-C2mim-1 and C-C4mim-1 complexes |
| 表 1 [C-C2/4mim+]作用体系在两种水平上的结合能(EB/EBSSE-B)和变形能(ED) Table 1 Binding energies(EB/EBSSE-B) and deforming energies(ED) of [C-C2/4mim+] at the M06-2X/6-311++G(2d, p) and ωB97XD/6-311++G(2d, p) levels |
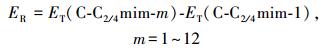
|
(1) |

|
(2) |
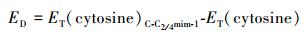
|
(3) |
式中, ET(C-C2/4mim-1)为C-C2/4mim-1的总能量, 变形能ED为C-C2/4mim-1结构中的碎片胞嘧啶结构作同水平的单点能计算, 再与自由状态下优化的胞嘧啶的单点能计算比较.下文[C-Br-]和[C-[C2/4mim]Br]作用体系各能量项定义与[C-C2/4mim+]体系一致.
胞嘧啶和C2mim+的作用方式有3种情况:第1种胞嘧啶与C2mim+共平面, 形成了最稳定结构(C-C2mim-1和C-C2mim-2), 其中胞嘧啶的C2=O7和阳离子的C2―H之间的距离在182.8~255.5 pm之间, 胞嘧啶的N3与阳离子的C2―H作用形成230.8~295.6 pm的氢键.以同种作用方式存在的C-C2mim-7、C-C2mim-8、C-C2mim-9、C-C2mim-11和C-C2mim-12结构中, 胞嘧啶主要作用于阳离子咪唑环的C4―H和C5―H, 得到的结构在能量学上不占优势.第2种情况是嘧啶环与咪唑环形成面垂直, 胞嘧啶的C2=O7作用于咪唑环的C4―H和C6―H, 且胞嘧啶的N3与咪唑环形成N3···π作用, 如结构C-C2mim-10.第3种存在于C-C2mim-4、C-C2mim-5和C-C2mim-6, 胞嘧啶作用于咪唑阳离子的上方, 嘧啶环与咪唑环近似垂直, 胞嘧啶的C2=O7和N3同时作用于阳离子的C2—H, 形成距离在186.8~253.1和222.9~302.4 pm范围内的氢键.胞嘧啶和C4mim+的作用方式与[C-C2mim+]体系相似, 其中胞嘧啶与C4mim+共平面, 形成最稳定结构C-C4mim-1, 在C-C4mim-1中C2—H···O7距离为201.1(193.8) pm.比较分析[C-C2/4mim+]体系中最稳定结构可知, C-C2mim-1中的C2—H···O7和C2—H···N3之间的距离比结构C-C4mim-1中的短约14.2 pm.
[C-C2/4mim+]体系中的胞嘧啶碎片与同水平优化的单体结构相比, 胞嘧啶碎片的C2=O7、N3=C4和C6—N1键伸长约12.0 pm, 胞嘧啶其余键长均缩短.气相中M06-2X方法计算C-C2/4mim-1中胞嘧啶碎片键长与ωB97XD方法计算结果相差约1.0 pm.
从图 3a可知, M06-2X/6-311++G(2d, p)计算气相中12种C-C2mim-m的相对能量ER相差在17.7 kJ· mol-1以内, M06-2X和ωB97XD两种水平计算同一结构得到的能量值相差小于5.8 kJ· mol-1, 图 3b中C-C4mim-m的相对能量ER相差在20.8 kJ· mol-1以内, 两种水平在气相中计算的C-C2mim-6的相对能量相差较大(12.3 kJ· mol-1), 其他结构相差不大.相对能差的结果表明C-C2mim-1和C-C4mim-1在[C-C2/4mim+]作用体系中最为稳定, 这与C2—H参与形成氢键有关, 由于咪唑阳离子的C2—H所带的正电荷最多, 因而形成的氢键键长最短, 氢键相互作用能最强, 与文献(吴阳等, 2009)中计算的1-乙基-3-甲基咪唑阳离子与天冬酰胺阴离子作用结果一致.后文重点考察最稳定结构C-C2/4mim-1的结合能和变形能, 数据列于表 1.气相中C-C2/4mim-1的结合能分别为-100.9(-105.7)和-101.7(-106.5) kJ· mol-1, 经过BSSE校正后, 结合能增加约2.0 kJ· mol-1. 图 2中C-C2/4mim-1结构中均存在四根氢键(C2—H···O7, C2—H···N3, C6—H···O7和C7—H···N3), 实验(Wu et al., 1999)测定的H—F···H—F体系中结合能为-18.65 kJ· mol-1, 这一结果表明胞嘧啶与阳离子之间的静电作用力较强.此外, 阳离子作用于胞嘧啶后, 其变形能为2.9(2.8)和3.3(3.1) kJ· mol-1, 胞嘧啶结构有所变化.
 |
| 图 3 [C-Cnmim+]作用体系在两种水平上经BSSE校正后的相对能量 Fig. 3 Relative energies of [C-C2/4mim+] at the M06-2X/6-311++G (2d, p) and ωB97XD/6-311++G(2d, p) levels |
考虑溶剂化效应后, 结构C-C2/4mim-1中的相互作用的原子间距离伸长约12.6 pm.水相中C-C2/4mim-1的相互作用能仅为-22.9(-26.9)和-24.9(-32.1) kJ· mol-1.与气相中的计算结果相比, 水相中的C-C2/4mim-1的相互作用能的大幅减小, 说明由于水溶液使胞嘧啶和C2/4mim+之间的静电作用减小, 表现为原子间距离的增加.胞嘧啶的变形能同样降低为0.5(0.4)和0.3(0.3) kJ· mol-1.表明水溶液降低了阳离子对胞嘧啶结构的影响.
2.1.2 [C-Br-]作用体系在M06-2X/6-311++G(2d, p)和ωB97XD/6-311++G (2d, p)水平计算的胞嘧啶与阴离子Br-作用体系中存在两种稳定结构, 如图 4所示.比较气相中两种水平计算的胞嘧啶与Br-相互作用原子间的距离发现ωB97XD方法计算的结果比M06-2X方法的结果键长短约1.2 pm.第1种结构(C-Br-1)存在N8—H···Br和C5—H···Br两种氢键类型, 形成的氢键键长分别为232.7(232.9)和284.3(290.6) pm. N8—H···Br键长比C5—H···Br的键长短, 是由于N8—H上所带的正电荷比C5—H的多0.165 e. C-Br-1结构中的N8—H···Br距离与文献(Tehrani et al., 2009)在B3LYP/6-311++G (d, p)基组水平计算得到的结构C-F-中的N8—H···F (109.1 pm)相比, 伸长123.6(123.8) pm; 与结构C-Cl-中的N8—H···Cl(211.1 pm)相比, 伸长21.6(21.8) pm, 这主要是源于卤素吸引电子的能力顺序为F>Cl>Br.此外, 复合物C-Br-1中胞嘧啶碎片与胞嘧啶单体结构相比, 胞嘧啶碎片的C2=O7、N3=C4、C4—C5和C6—N1键长伸长约1.2 pm, 胞嘧啶其余键长缩短约1.2 pm, 这些变化均由Br-向N8—H和C5—H的空轨道提供电子而引起环上电子流动所致.第2种作用方式(C-Br-2)为溴离子与胞嘧啶的N1—H和C6—H作用, 键长分别为246.7(248.6)和273.4(277.1) pm. ωB97XD方法计算的相互作用原子间距离比M06-2X方法计算的伸长约2.8 pm.
 |
| 图 4 胞嘧啶与阴离子(Br-)复合物的几何结构 Fig. 4 Geometrical structures of [C-Br-] complexes |
气相中M06-2X和ωB97XD方法计算的两种结构的能量学数据列入表 2中, 其相对能量相差12.0(13.4) kJ· mol-1, 两种稳定结构的结合能分别为-102.2(-99.3)和-90.2(-85.9) kJ· mol-1.经BSSE校正之后其结合能降低约1.2 kJ· mol-1.由此可以得到两种结构的稳定性顺序为C-Br-1>C-Br-2.在C-Br-1结构中, 胞嘧啶的变形能为5.3(5.1) kJ· mol-1, 与C-C2/4mim-1中的胞嘧啶变形能相比增加2.4(2.3) kJ· mol-1, 变形能增大意味着嘧啶环平面性差, 环原子共轭效应减弱, 引起结构稳定性降低.
| 表 2 [C-Br-]作用体系在两种水平上的相对能(ER)、结合能(EB/EBSSE-B)、变形能(ED) Table 2 Relative energies(ER), binding energies(EB/EBSSE-B), deforming energies(ED) of [C-Br-] complex at the levels of M06-2X/6-311++G(2d, p) and ωB97XD/6-311++G(2d, p) |
在相同的计算水平下, 采用PCM模型对其水相中的结构进行研究, 得到的几何结构参数以红色斜体标示在图 4中.以C-Br-1为例, 其C5—H···Br和N8—H···Br之间的距离明显伸长, 氢键作用减弱, 水相中C-Br-1经BSSE校正后的结合能能仅为-16.6(-15.6) kJ· mol-1, 表明水溶液不利于[C-Br-]体系的稳定.在水溶液中胞嘧啶的变形能仅为0.6(0.3) kJ· mol-1, 表明在水溶液中溴离子对胞嘧啶的结构影响不大.
2.1.3 [C-[C2/4mim]Br]作用体系在M06-2X/6-311++G(2d, p)和ωB97XD/6-311+G(2d, p)水平上优化得到[C-[C2mim]Br]和[C-[C4mim]Br]体系的31种稳定结构, [C-[C2/4mim]Br]体系中最稳定结构并不是由最稳定的C-C2/4mim-1或C-Br-1形成的.以M06-2X/6-311++G(2d, p)水平上计算的电子相对能量ER由低到高依次编号C2/4m(m=1~31).胞嘧啶与[C2mim]Br的作用方式可分为3种类型.第1种为共平面结构(如C220-C224), 但其具有较高的能量, 热力学上不稳定.第2种为胞嘧啶和阳离子咪唑环近似平行, 阴离子作用于咪唑环的C2—H和胞嘧啶的N1—H(如C212), 嘧啶环和咪唑环之间可能存在π-π相互作用.第3种作用方式为嘧啶环和咪唑环接近垂直, 其结构能量较低, 是胞嘧啶和[C2mim]Br之间的主要作用方式, 如在C21中, 阴离子位于咪唑环的平面上方, 胞嘧啶垂直于C2—H键和阴离子, 胞嘧啶的N1—H和阴离子作用, N1—H···Br距离为223.8(223.6) pm, 咪唑环的C2—H与胞嘧啶的氧原子(C2=O7)作用形成氢键距离为203.0(196.4) pm.胞嘧啶的C2=O7氢键受体可以与C2—H形成稳定的氢键, 这与咪唑环上的电子缺失导致C2—H成为咪唑环上带最多正电荷的氢原子是一致的. C41为[C-[C4mim]Br]体系中最稳定的结构, 与C21相比, 其C2—H···O7之间的距离缩短18.0(18.0) pm.在复合物C2/41中胞嘧啶碎片与胞嘧啶单体结构相比, 其C2=O7、N3—C4、C5=C6和C4—N8键长伸长约6.0 pm, 胞嘧啶其余键长缩短约8.0 pm.
图 5为[C-[C2/4mim]Br]体系在两种水平计算得到的31种结构的相对能量, 在M06-2X和ωB97XD水平上31种[C-[C2mim]Br]结构的相对能量相差82.9(80.7) kJ· mol-1.经过基组重叠误差校正后[C-[C2mim]Br]的相对能量为81.0(78.9) kJ· mol-1.比较发现[C-[C2mim]Br]和[C-[C4mim]Br]之间的相对能差无显著差异.此外, 图 6中的结构C21和C41为[C-[C2/4mim]Br]作用体系(除水相中的结构C42和C49)中最稳定的结构.对C21结构中C2—H···Br和C2—H···O7作用位做势能曲线, 其键长每10.0 pm变化1次.结果表明在气相中, 随着C2—H···Br和C2—H···O7键长的增加, 体系的相对能量逐渐减小, 在键长为340.3(335.8)和203.0(196.2) pm时能量最低.势能曲线的最低点与M06-2X和ωB97XD方法计算得到的最稳定结构中C2—H···Br和C2—H···O7键长一致. C21和C41的结合能和变形能的数据列于表 3, C21和C41的结合能分别为-101.9(-105.7)和-109.7(-111.3) kJ· mol-1, 经BSSE校正后, 其结合能降低约5.1 kJ· mol-1.气相中C21和C41的变形能为6.5(6.9)和6.2(6.9) kJ· mol-1, 与[C-C2/4mim+]、[C-Br-]体系最稳定结构中胞嘧啶的变形能相比明显增大, 表明阴阳离子作用于胞嘧啶时, 其结构变化最大.
 |
| 图 5 [C-[C2/4mim]Br]作用体系经BSSE校正后的相对能量 Fig. 5 Relative energies are corrected by the basis set superposition error(BSSE) of [C-[C2/4mim]Br] complexes |
 |
| 图 6 C21和C41的几何结构 Fig. 6 Geometrical structures of C21 and C41 complexes |
| 表 3 [C-[C2/4mim]Br]作用体系中最稳定结构在两种水平上的结合能(EB/EBSSE-B)和变形能(ED) Table 3 Binding energies(EB/EBSSE-B) and deforming energies(ED) of C2/41 at the M06-2X/6-311++G(2d, p) and ωB97XD/6-311++G(2d, p) levels |
比较气相和水相中的C21和C41的几何结构发现, 水溶液使相互作用的原子(C2—H···O7和N1—H···Br)之间的距离伸长了约7.8和14.7 pm.同时在水溶液中C21和C41经BSSE校正后的结合能仅为-32.7(-36.8)和-36.3(-39.1) kJ· mol-1, 与气相相比降低了约65.8 kJ· mol-1, 胞嘧啶的变形也仅为1.1(1.4)和1.2(1.6) kJ· mol-1. 3种作用体系中最稳定结构的几何结构、结合能和变形能均表明水溶液环境能减轻C2/4mim+、Br-和[C2/4mim]Br对胞嘧啶的影响.
2.2 谱学性质采用M06-2X和ωB97XD方法在6-311++G(2d, p)基组水平下对Cytosine、[C-C2/4mim+]、[C-Br-]和[C-[C2/4mim]Br]体系最稳定结构的拉曼光谱进行计算, 理论计算的胞嘧啶拉曼振动光谱振动强度较大的峰主要集中在1300~1700和3200~3800 cm-1之间, 胞嘧啶中的最强峰为—NH2的伸缩振动, 位于3624(3635) cm-1处.此外, 位于1816(1805) cm-1处的吸收峰是C2=O7的伸缩振动, 与Madzharova等(2016)在表面增强拉曼光谱(SEHRS)中观察到胞嘧啶位于1635 cm-1处的C2=O7伸缩振动之间的差异较大, 主要是因为理论计算的是单个胞嘧啶分子在气相或水相中的结构参数, 而实验中的结构参数是由胞嘧啶晶体在固相中获得的.
为了比较C2/4mim+、Br-和[C2/4mim]Br对体系中胞嘧啶形成作用中心键的伸缩振动频率(v)变化的影响, 按式(4)计算了在M06-2X/6-311++G(2d, p)水平得到的3种体系中最稳定结构作用中心键的振动频率差值Δv, 见图 7.
 |
| 图 7 [C-C2/4mim+]、[C-Br-]和[C-[C2/4mim]Br]体系中胞嘧啶作用中心键的伸缩振动频率差值图 Fig. 7 Difference values of stretching vibrational frequencies of interaction center bonds of cytosine |
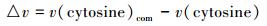
|
(4) |
在[C-C2/4mim+]和[C-[C2/4mim]Br]体系中, C-C2/4mim-1和C2/41结构的vC2=O7红移约62 cm-1, 且在C2/41中存在的阴离子Br-作用于胞嘧啶的N1—H使C2=O7键的红移程度比C-C2/4mim-1中的大.结构C2/41中的N1—H同样发生红移, 其振动变化约为498 cm-1. [C-C2/4mim+]体系中的C2—N3键发生蓝移, 振动变化约34 cm-1.在[C-Br-]体系中的vN8—H红移约403 cm-1, vC5—H键红移37(21) cm-1.采用PCM模型考虑了水溶液环境后, C-C2/4mim-1、C-Br-1和C2/41结构中键的振动变化趋势与气相中一致.例如C-Br-1中的vN8—H红移了162(207) cm-1, 结构C21中C2=O7键吸收峰红移了12(6) cm-1.对比气液两相中的计算结果, 发现水相中的红移明显减小, 可见水相状态下[C-C2/4mim+]、[C-Br-]和[C-[C2/4mim]Br]中相互作用明显减弱.
2.3 分子中的原子理论Bader的AIM分析被认为是判断氢键存在及其强弱的有力工具, 已经广泛应用到对各种氢键的讨论中.其中键临界点(BCP)处的电荷密度ρBCP用来表示原子间成键的强弱, 电荷密度越大, 表明该处的氢键作用越强, 反之氢键作用越弱.电子云密度的拉普拉斯值∇2ρBCP和键临界点处的HBCP可用来判断原子间相互作用的性质, 若∇2ρBCP和HBCP的值为正值, 表明原子间的作用以静电作用为主; 若∇2ρ和HBCP的值为负值, 表明以共价键为主.
在M06-2X/6-311++G(2d, p)水平下对3种体系进行了AIM计算,得到5种最稳定结构在BCP处的电子密度ρBCP、拉普拉斯值∇2ρBCP以及电子能量密度HBCP.计算结果表明气相中胞嘧啶与C2/4mim+、Br-和[C2/4mim]Br的电子密度ρBCP分别在0.005~0.026、0.010~0.024和0.007~0.029 a.u.之间, 与传统的氢键电子密度(0.002~0.040 a.u.)相近.在C-C2/4mim-1中C2—H···O7的氢键作用最强, 在C-Br-1中N8—H···Br的氢键作用最强, 但当阴阳离子同时作用于胞嘧啶时, 复合物中N1—H···Br和C2—H···O7的氢键作用最强, 但C2—H···O7的强度比阳离子作用于胞嘧啶时弱. 3个相互作用体系计算得到的电子云密度的拉普拉斯值∇2ρBCP全部为正值, 即复合物体系的氢键结构中静电作用占主导地位.在C21和C41结构中的N1—H···Br之间相互作用的电子能量密度HBCP为负, 表明其在非键作用有部分的共价性.
在相同水平水溶液中的AIM分析表明胞嘧啶与C2/4mim+的作用体系中电子密度ρBCP最高值为0.019, 拉普拉斯值∇2ρBCP全部为正值, 但与气相中相比明显降低. C-Br-1中的N8—H···Br和C5—H···Br的电子密度及拉普拉斯值同样与气相中的数值相比降低了约0.004和0.095 a.u.. C21和C41中的电子能量密度HBCP全部为正值, 说明在水相中其静电相互作用仍为主要的性质.
 |
| 图 8 在M06-2X/6-311++G(2d, p)水平计算气相中C-C2/4mim-1、C-Br-1和C2/41的分子图 Fig. 8 AIM molecular graph showing the different bond critical points(BCPs) and bond paths of C-C2/4mim-1, C-Br-1 and C2/41 |
在M06-2X/6-311++G(2d, p)水平下[C-C2/4mim+]、[C-Br-]和[C-[C2/4mim]Br]体系最稳定结构自然键轨道布分析的碎片所带电荷,列入表 4中,原子标号见图 1.气相中, C-C2/4mim-1的胞嘧啶和C2/4mim+碎片所带电荷为0.038和0.962 e, C-C2mim-1和C-C4mim-1的电荷转移量相差不大, 约为0.037 e.当溴离子作用于胞嘧啶时, 两者之间的电荷转移量为0.070 e, 与C-C2/4mim-1相比, C-Br-1之间的电荷转移更多, 形成的氢键作用更强.在复合物C21中, 胞嘧啶、C2mim+和Br-的转移量分别为-0.061、0.055和0.006e和-0.057、0.049和0.008e, 表明当[C2mim]Br作用于胞嘧啶时, 体系中的电荷转移主要发生在C2mim+向胞嘧啶转移.考虑溶剂化效应后, [C-C2/4mim+]和[C-Br-]体系的电荷转移量减少约0.018 e, [C-[C2/4mim]Br]体系中, 阴离子Br-向胞嘧啶和C2/4mim+转移了约0.029和0.015 e.
| 表 4 在M06-2X/6-311++G(2d, p)方法下C-C2/4mim-1、C-Br-1和C2/41结构的二阶稳定化能E(2) Table 4 The second-order interactions energies E(2) of the C-C2/4mim-1, C-Br-1 and C2/41 at the M06-2X/6-311++G(2d, p) level |
为了研究[C-C2/4mim+]、[C-Br-]和[C-[C2/4mim]Br]这3种体系分子间相互作用本质, 在M06-2X/6-311++G(2d, p)水平下对3个体系进行了自然键轨道分析.根据文献(Qu et al., 2012)对于稳定化能的筛选标准(E(2)>2 kJ· mol-1), 表 4列出了最稳定结构中电子供体轨道i和电子受体轨道j相互作用的二阶稳定化能E(2), 其值越大表明i与j的相互作用越强, 氢键作用越强.式(5)中, qi是供体轨道占据的电荷数, εi和εj是轨道能量, F(i, j)是Fock矩阵的矩阵元.
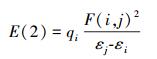
|
(5) |
在C-C2mim-1和C-C4mim-1中, 胞嘧啶中C2=O7的孤对电子和C2/4mim+中的C2—H反键轨道的相互作用最大, 其稳定化能分别为13.64和11.80 kcal· mol-1, 表明C-C2/4mim-1的氢键稳定化能主要来源于LP O7→BD*C2—H.在C-Br-1中, 其稳定化能主要来源于阴离子的孤对电子和胞嘧啶N8—H反键轨道之间的相互作用(LP Br→BD*N8—H), 其稳定化能为11.80 kcal· mol-1.对比图 2和图 6中相互作用原子间的距离可知, 其相互作用距离越短, 稳定化能越高.从C2/41的计算结果可以看出, 胞嘧啶与离子液体的稳定化能主要来源于LP Br→BD*N1—H、LP O7→BD*C2—H和LP Br→BD*N1—C2.
3 结论(Conclusions)利用M06-2X/6-311++G(2d, p)和ωB97XD/6-311++ G(2d, p)方法计算了咪唑阳离子、溴阴离子和溴化咪唑类离子液体对胞嘧啶结构和性质的影响, 得到了2种[C-Br-]、12种[C-C2/4mim+]和31种[C-[C2/4mim]Br]稳定结构, 详细讨论了其静电势、几何结构、电子结构、能量学特征和谱学特征, 可以得到以下几点结论:①在[C-[C2/4mim]Br]体系中最稳定结构并不是由[C-C2/4mim+]或[C-Br-]体系中最稳定的结构形成的, 且胞嘧啶在[C-[C2/4mim]Br]体系中结构变化最大. ②3种体系中最稳定的结构分别为C-C2/4mim+-1、C-Br-1和C2/41, 经BSSE校正后结合能分别在-99.1~-104.3、-98.0~-101.2和-97.3~-105.6 kJ· mol-1范围内. ③当C2/4mim+、Br-和[C2/4mim]Br作用于胞嘧啶时, 胞嘧啶C2=O7和N1—H键的伸缩振动频率发生一定程度的红移. ④AIM和NBO分析发现, 在C2/41结构中氢键的形成来自电子供体的溴阴离子所带的孤对电子向咪唑阳离子上C2—H的反键轨道转移的离域效应和胞嘧啶上C2=O7的孤对电子向咪唑阳离子上C2—H的作用. ⑤气相与液相计算结果的对比表明, 极性溶剂环境削弱了3种体系中的静电作用, 致使C2/4mim+、Br-和[C2/4mim]Br对胞嘧啶的作用相对较弱.
Bader R F W. 1990. Atom in molecular-A quantum theory[J]. Computer Chemical, 4: 1.
|
Chai J D, Head G M. 2008. Optimal operators for Hartree-Fock exchange from long-range corrected hybrid density functionals[J]. Chemical Physical Letter, 467: 176.
|
Chai J D, Head G M. 2009. Long-range corrected double-hybrid density functionals[J]. The Journal of Chemical Physics, 131(17): 174105.
DOI:10.1063/1.3244209
|
Chandran A, Ghoshdastidar D, Senapati S. 2012. Groove binding mechanism of ionic liquids:A key factor in long-term stability of DNA in hydrated ionic liquids?[J]. Journal of the American Chemical Society, 134(50): 20330.
DOI:10.1021/ja304519d
|
Costa S P F, Azevedo A M O, Pinto P, et al. 2017. Environmental impact of ionic liquids:an overview of recent (eco)toxicological and (bio)degradability literature[J]. Chem Sus Chem, 10(11): 2321.
|
Ding Y, Zhang L, Xie J. 2010. Binding characteristics and molecular mechanism of interaction between ionic liquid and DNA[J]. Journal of Physical Chemistry B, 114(5): 2033.
DOI:10.1021/jp9104757
|
Dreyfus M, Bensaude O, Dodin G, et al. 1976. Tautomerism in cytosine and 3-methylcytosine. A thermodynamic and kinetic study[J]. Journal of the American Chemical Society, 98(20): 6338.
|
Du Z, Zhu L, Dong M. 2012. Effects of the ionic liquid[Omim]PF6 on antioxidant enzyme systems, ROS and DNA damage in zebrafish (Danio rerio)[J]. Aquatic Toxicology, 124: 91–93.
|
Frisch M J, Trucks G W, Schlegel H B, et al. 2013. Gaussian 09, Revision D.01. Gaussian, Inc., Wallingford CT
|
Grimme S. 2011. Density functional theory with London dispersion corrections[J]. Wiley Interdisciplinary Reviews Computational Molecular Science, 1(2): 211–228.
|
Gutiérrez A, Atilhan M, Alcalde R. 2018. Insights on the mixtures of imidazolium based ionic liquids with molecular solvents[J]. Journal of Molecular Liquids, 255: 199.
|
Jumbri K, Abdul R M B, Abdulmalek E. 2014. An insight into structure and stability of DNA in ionic liquids from molecular dynamics simulation and experimental studies[J]. Physical Chemistry Chemical Physics, 16(27): 14036–14046.
|
井长勤, 陈红丽, 李效宇. 2013. 离子液体氯化1-辛基-3-甲基咪唑对EMT6细胞的毒性及其机理研究[J]. 环境科学学报, 2013, 33(6): 1809–1814.
|
Khan A S, Man Z, Arvina A, et al. 2017. Dicationic imidazolium based ionic liquids:synthesis and properties[J]. Journal of Molecular Liquids, 227: 98.
|
Liu H J, Xia Y L, Cai W D. 2017. Enantioselective oxidative stress and oxidative damage caused by Rac-and S-metolachlor to Scenedesmus obliquus[J]. Chemosphere, 173: 22–30.
DOI:10.1016/j.chemosphere.2017.01.028
|
Liu H J, Zhang S X, Zhang X Q. 2015. Growth inhibition and effect on photosystem by three imidazoliumchloride ionic liquids in rice seedlings[J]. Journal of Hazardous Materials, 286: 440.
DOI:10.1016/j.jhazmat.2015.01.008
|
Lu T, Chen F. 2012. Multiwfn:A multifunctional wavefunction analyzer[J]. Journal of Computational Chemistry, 33(5): 580–592.
|
Madzharova F, Heiner Z, Gühlke M, et al. 2016. Surface-enhanced hyper-raman spectra of adenine, guanine, cytosine, thymine, and uracil[J]. Journal of Physical Chemistry C, 120: 15415.
DOI:10.1021/acs.jpcc.6b02753
|
Nancharaiah Y V, Francis A J. 2011. Alkyl-methylimidazolium ionic liquids affect the growth and fermentative metabolism of Clostridium sp.[J]. Bioresource Technology, 102(11): 6573–6578.
DOI:10.1016/j.biortech.2011.03.042
|
Orozco M, Luque F J. 2000. Theoretical methods for the description of the solvent effect in biomolecular systems[J]. Chemical Reviews, 100(11): 4187–4226.
DOI:10.1021/cr990052a
|
Qu R J, Liu H X, Feng M B, et al. 2012. Investigation on intramolecular hydrogen bond and some thermodynamic properties of polyhydroxylated anthraquinones[J]. Journal of Chemical & Engineering Data, 57(9): 2442–2455.
|
Steudte S, Stepnowski P, Cho C W, et al. 2012. (Eco)toxicity of fluoro-organic and cyano-based ionic liquid anions[J]. Chemical Communications, 48(75): 9382–9384.
DOI:10.1039/c2cc34955h
|
Tehrani Z A, Fattahi A. 2009. Anion interactions of cytosine nucleobase and its nucleosides:Detailed view from DFT study[J]. Journal of Molecular Structure Theochem, 913(1): 277–283.
|
Tomic K, Tatchen J R, Marian C M. 2005. Quantum chemical investigation of the electronic spectra of the keto, enol, and keto-imine tautomers of cytosine[J]. Journal of Physical Chemistry A, 109(37): 8410.
DOI:10.1021/jp051510o
|
Ueda T, Fox J J. 1963. Spectrophotometric studies of nucleic acid derivatives and related compounds. V. on the structure of 3-methylcytosine[J]. Journal of the American Chemical Society, 85: 4024.
|
Vasiliy S Z, Michael E G. 2004. Topological changes of hydrogen bonding of water with acetic acid:AIM and NBO studies[J]. Journal of Physical Chemistry A, 108(31): 6543–6553.
|
Vekariya R L. 2017. A review of ionic liquids:Applications towards catalytic organic transformations[J]. Journal of Molecular Liquids, 227: 44.
DOI:10.1016/j.molliq.2016.11.123
|
Ventura S P M, Neves C M, Freire M G. 2009. Evaluation of anion influence on the formation and extraction capacity of ionic-liquid-based aqueous biphasic systems[J]. Journal of Physical Chemistry B, 113(27): 9304–9310.
|
Wang B, Qin L, Mu T, et al. 2017. Are ionic liquids chemically stable?[J]. Chemical Reviews, 117(10): 7113–7131.
DOI:10.1021/acs.chemrev.6b00594
|
Wu X T, Hayes E F, Mccoy A B. 1999. Rotation-vibration interactions in HF2. Ⅱ. Rotation-vibration interactions in low-lying vibrational states[J]. Journal of Chemical Physical, 110: 2365–2370.
|
吴阳, 岳丽丽, 刘巧珍, 等. 2016. 咪唑醋酸离子液体热力学性质的理论研究[J]. 物理化学学报, 2016, 32: 1960.
DOI:10.3866/PKU.WHXB201605123 |
吴阳, 张甜甜, 于宁. 2009. 1-乙基-3-甲基咪唑阳离子与天冬酰胺阴离子的相互作用[J]. 物理化学学报, 2009, 25: 1689.
DOI:10.3866/PKU.WHXB20090823 |
张瑾, 刘树深, 王成林, 等. 2013. 离子液体与废水对青海弧菌Q67的混合毒性研究[J]. 环境科学学报, 2013, 33(3): 850–855.
|