 2018, Vol. 38
2018, Vol. 38



2. 挥发性有机物污染治理技术与装备国家工程实验室, 广州 510006;
3. 广东省大气环境与污染控制重点实验室, 广州 510006;
4. 广东省环境风险防控与应急处置工程技术研究中心, 广州 510006
2. National Engineering Laboratory for VOCs Pollution Control Technology and Equipment, Guangzhou 510006;
3. Guangdong Provincial Key Laboratory of Atmospheric Environment and Pollution Control(SCUT), Guangzhou 510006;
4. Guangdong Provincial Engineering and Technology Research Centre for Environmental Risk Prevention and Emergency Disposal, South China University of Technology, Guangzhou 510006
近年来, 挥发性有机物(Volatile organic compounds)的污染治理受到高度重视.催化燃烧法由于具有高去除效率、无二次污染等优势, 被认为是治理VOCs最有效的技术之一(Genuino et al., 2012), 而研发高效的催化剂是提升该技术核心竞争力的关键.贵金属催化剂, 尤其是铂基催化剂, 对于VOCs的催化燃烧具有较高的催化活性, 被广泛应用于众多行业VOCs的净化.而载体的选择对于其催化性能具有重要影响, 其中, 稀土氧化物CeO2作为典型的活性载体, 具有优异的储放氧能力.CeO2纳米材料一方面可以锚定贵金属颗粒, 提升催化剂稳定性;另一方面可以直接参与反应, 提供新的反应活性位点, 进而提升反应性能.近年来, 许多科研工作者围绕Pt/CeO2催化剂的制备及其性能开展了大量研究.例如, Gao等(2013)研究了CeO2载体形貌对Pt/CeO2催化氧化CO反应的影响, 发现Pt/r-CeO2展现出最好的催化性能; Lee等(2016)制备了具有核壳结构的Pt@CeO2纳米材料, 提高了催化燃烧甲烷的活性及热稳定性; 本课题组在Pt/CeO2催化材料结构和性能研究方面也取得了一定成果, 发现载体形貌及Pt颗粒尺寸对催化氧化甲苯性能有重要影响(Peng et al., 2016).
在VOCs的热催化机理研究方面, 近年来科研工作者采用先进的原位手段, 针对催化剂反应活性位点及反应过程进行了更深入的研究, 为高效催化剂的研发提供了理论依据.Zhang等(2006)通过原位DRIFTS技术研究了Pt/TiO2在室温下催化氧化甲醛的反应过程; Moreno-González等(2014)利用原位EPR和IR技术研究了Cu-zeolites催化氧化丙烷的反应活性位点, 发现吸附在材料上的COO—和—CHO是丙烷反应中间物种;Lai等(2014)通过DFT理论计算与In-situ DRIFTS研究了Pt/SBA-15和Pt/SiO2催化剂上Pt原子的配位情况及CO、甲苯吸附过程.由于VOCs污染物种类繁多且结构复杂, 不同催化体系下的研究成果不尽相同.目前, 针对Pt/CeO2体系催化氧化甲苯的反应机制研究还不是很广泛.
本研究通过浸渍法制备Pt/CeO2、Pt/Al2O3催化剂, 采用H2-TPR、甲苯-TPD、GC/MS及In-situ FTIR等技术对比研究两种催化剂的物理化学性能、甲苯吸附-脱附及反应中间产物/副产物.
2 实验部分(Experiment part) 2.1 催化剂制备载体制备:CeO2纳米棒制备方法参考前期研究(廖银念, 2013), 以醋酸铈(Ce(CH3COO)3·nH2O)为铈源, 利用水热合成法制备而得, 记为CeO2; γ-Al2O3选用商业氧化铝作为载体(阿法埃莎(中国)化学有限公司).
Pt/CeO2及Pt/Al2O3催化剂的制备:采用湿浸渍法, 分别将相应Pt含量(0.6%, 质量分数)的硝酸铂溶液加入到CeO2和γ-Al2O3悬浊液中, 在恒温(40 ℃)摇床上振荡过夜, 直至样品干燥; 再将所得粉末转移至马弗炉中400 ℃焙烧4 h; 产物于5% H2/N2气氛下, 300 ℃还原处理3 h, 最终获得对应的催化剂.
2.2 催化剂表征X-射线衍射分析(XRD):使用德国Bruker D8 Advance X衍射仪, 测试使用Cu Kα靶, 入射波长λ=0.15418 nm, 扫描范围2θ=10°~90°, 扫描速度为6°·min-1.
高分辨透射电子显微镜(TEM&HRTEM):使用日本JEOL JEM-2100HR型透射电子显微镜, 样品观察前进行超声处理.
程序升温还原(H2-TPR):使用美国Micromeritics AutoChem 2920全自动程序升温化学吸附仪进行测定, 测试前将样品在5% O2/He(30 mL·min-1)气氛下, 300 ℃加热1 h, 在氩气(30 mL·min-1)环境下将样品降至室温; 测试时使用10% H2/Ar(30 mL·min-1), 待基线稳定后, 以10 ℃·min-1升温速率从室温升到800 ℃.
比表面积测试(BET):使用美国Micromeritics公司的ASAP 2020 M全自动表面分析仪, 以N2为吸附质测定, 测试前300 ℃抽真空处理4 h, 测试温度为-196 ℃.
电感耦合等离子体原子吸收分析(ICP-OES):使用美国Perkin-Elmer plasma 8000型ICP-OES仪测定.
CO脉冲吸附:利用Micromeritics AutoChem 2920测试Pt颗粒分散度和粒径, 计算设定Pt:CO的化学计量比为1, 原子断面面积为0.0787 nm2. Pt颗粒粒径d(nm)的计算公式如下:
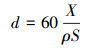
|
(1) |
式中, X为Pt的负载量, ρ为Pt的密度(g·cm-3), S为Pt的比表面积(m2).
X-射线光电子能谱(XPS):使用美国Thermo-VG Scientific ESCALAB 250光电子能谱仪进行测定, 测试使用Mg Ka (h=1253.6 eV)射线, C1s校准结合能为284.6 eV.
拉曼光谱分析(Raman):使用法国HYJ公司的LabRAM Aramis型拉曼光谱仪进行测定, 使用325 nm UV光源, 扫描范围为300~1400 cm-1.
2.3 催化剂活性评价将200 mg 40~60目的催化剂与800 mg石英砂混合均匀装入内径为8 nm的石英管内, 通入甲苯与干燥空气混合气进行甲苯氧化反应(甲苯浓度为1000 ppm, 流量160 mL·min-1, 重时空速48000 mL·g-1·h-1).反应前后的气体均由气相色谱仪(GC-2014C, 日本)进行检测分析, 测得反应前后甲苯浓度.甲苯转化率(η)计算公式如下:
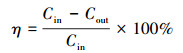
|
(2) |
式中, Cin为甲苯进口浓度, Cout为甲苯出口浓度.
2.4 反应过程分析甲苯TPD:将200 mg 40~60目催化剂置于固定床内, 在室温条件下通入浓度为200 ppm甲苯/N2(70 mL·min-1), 直至吸附饱和.再用N2(70 mL·min-1)吹扫60 min, 并在N2气氛下以一定升温速率(3 ℃·min-1)升温到230 ℃. O2预吸附处理的催化剂(分别记为O2-Pt/CeO2、O2-Pt/Al2O3)先在室温通入干燥空气处理30 min, N2吹扫60 min之后, 再进行甲苯TPD测试.尾气通入Hidden HPR-20质谱仪进行甲苯、H2、CO2信号在线检测, 其中, 在进行CO2信号测试时, 保持在甲苯气氛中升温.
原位红外透射光谱(In-situ FTIR):使用美国热电公司Nicolet 6700傅里叶变换红外光谱仪, 扫描范围为650~4000 cm-1, 扫描次数16次, 分辨率4 cm-1.
气态副产物检测:在一定反应温度下, 使用吸附管(Carbon trap)吸附包含气态副产物的反应尾气30 min, 将样品进行热脱附处理(PerkinElmer TurboMatrix 350), 脱附气体接入日本岛津公司QP2010 GC/MS进行成分分析.
3 结果与讨论(Results and discussion) 3.1 材料表征Pt/CeO2及Pt/Al2O3的XRD图谱如图 1所示.图中Pt/CeO2出现的特征峰位与CeO2立方萤石结构的标准卡片吻合(JCP 34-0394), Pt/Al2O3样品的衍射峰则归属于γ-Al2O3的特征峰(JCPDS 29-0063).此外, 两个样品并没有出现与Pt相关的衍射峰(33°、44°), 这可能是由于Pt的负载量较低, 另一方面可能因为Pt颗粒较好地分散在载体表面(Huang et al., 2014).
 |
| 图 1 Pt/CeO2和Pt/Al2O3的XRD谱图 Fig. 1 The XRD pattern of Pt/CeO2 and Pt/Al2O3 |
图 2为催化剂Pt/CeO2和Pt/Al2O3的TEM图和HRTEM图.从图 2a、2b中可以看出, CeO2呈现良好的棒状形貌, 其长度为100~200 nm, 宽为5~10 nm, 且暴露出(110)、(200)、(111)晶面, 而Pt/Al2O3样品并未展现特定形貌(图 2c、2d).此外, 在两个样品上均发现了Pt纳米颗粒的存在, 都具有(200)晶面.
 |
| 图 2 Pt/CeO2 TEM (a)、Pt/CeO2 HRTEM (b)、Pt/Al2O3 TEM (c)及Pt/Al2O3 HRTEM (d)图 Fig. 2 The TEM image of Pt/CeO2(a), Pt/Al2O3(c), and partial HRTEM of Pt/CeO2(b) and Pt/Al2O3(d) |
表 1列出了各样品的比表面积、Pt负载量、Pt尺寸及其分散度, 其中, 载体在进行比表面积测试前进行了400 ℃焙烧处理.可以看出, Pt/CeO2和Pt/Al2O3样品的比表面积较其载体略微减小, 这是由于贵金属负载会堵塞部分载体材料的微孔, 也可能是因为高温焙烧处理导致催化剂局部烧结.两样品的Pt负载量分别为0.61%和0.60%, 相差不大.CO脉冲吸附实验结果表明, 催化剂Pt/CeO2和Pt/Al2O3的Pt颗粒尺寸分别为2.5和2.2 nm, 分散度分别为0.45和0.50.相比之下, Pt/Al2O3的Pt颗粒尺寸更小, 分散度更高, 主要是因为γ-Al2O3载体拥有更大的比表面积(259.8 m2·g-1>102.6 m2·g-1), 通过浸渍法负载的Pt纳米颗粒在载体表面分散得更好.
| 表 1 不同样品的结构参数 Table 1 Textural parameters of various samples |
样品的可还原性能通过H2-TPR测定, 结果如图 3a所示.作为典型的惰性载体, γ-Al2O3并没有出现还原峰, 而Pt/Al2O3在100 ℃左右出现一个较弱的还原峰, 属于表面PtOx的还原峰(Borgna et al., 1992).而CeO2样品在300 ℃和428 ℃出现还原峰, 分别归属于表面氧及次表层氧的还原(López et al., 2015).负载Pt后的Pt/CeO2的图谱发生了较大变化, 在92 ℃出现了一个新的还原峰, 该峰归属于表面PtOx的还原(Reyes et al., 1997); 另外, 由于溢出效应, 该峰还包含了与Pt颗粒相邻的Ce4+的还原(Liu et al., 2014).值得关注的是, 与纯CeO2相比, 其CeO2表面氧的还原峰(250 ℃)向低温段发生较大偏移且耗氢量更高, 为Pt/CeO2(1137 μmol·g-1)>CeO2(531 μmol·g-1).这是由于Pt与CeO2之间的相互作用, 使得与Pt相邻的Ce—O键更容易断裂(Rui et al., 2008; Huang et al., 2014), 增强了CeO2载体表面氧的溢出(Vayssilov et al., 2011), 提高了Pt/CeO2催化剂的可还原性.
 |
| 图 3 Pt/CeO2、CeO2、Pt/Al2O3和γ-Al2O3的H2-TPR谱图(a)及Pt/CeO2、CeO2的UV Raman谱图(b) Fig. 3 The H2-TPR pattern of Pt/CeO2, CeO2, Pt/Al2O3 and γ-Al2O3(a) and the UV Raman pattern of Pt/CeO2 and CeO2(b) |
对于铈基催化剂, 表面氧空位是氧化反应中一个重要影响因素(Wu et al., 2010; Calaza et al., 2012).图 3b为Pt/CeO2和CeO2材料的紫外拉曼谱图, 其中, 458 cm-1处是CeO2立方萤石结构的对称收缩振动特征峰(F2g) (Weber et al., 1993); 592 cm-1处是因为Ce3+存在而产生的表面氧空位的特征峰(D)(McBride et al., 1994); 而417 cm-1处的峰在其他CeO2纳米棒的拉曼图谱中也有出现, 其归属暂不清晰(Agarwal et al., 2014).通常用D峰和F2g峰的强度比值(ID/IF2g)来表征CeO2氧空位的相对浓度.结果表明, 负载Pt之后, CeO2氧空位浓度显著增加, 为Pt/CeO2(2.102)>CeO2(1.493), 这可能由于Pt活化了CeO2表面氧及晶格氧, 降低了CeO2氧空位形成能(Wang et al., 2015; Chang et al., 2012).
通过XPS测试可以获得样品表面元素化学价态信息.图 4为各个催化剂Pt 4f、Al 2p、Ce 3d的XPS谱图.对于Pt/CeO2催化剂, Pt 4f范围的包峰可以分峰为71.6、73.1、74.3及75.8 eV 4个峰, 其中, 位于71.6和74.3 eV处的峰归属于Pt0物种, 余下的73.1和75.8 eV峰代表Pt2+物种(Gao et al., 2013).如表 2所示, Pt在表面主要以金属态形式存在, Pt0/(Pt2++Pt0)占比达到70%以上; 而Pt/Al2O3催化剂, 由于Pt 4f与Al 2p的特征峰位重叠, 无法判断Pt的价态.另外, 如Ce 3d的XPS谱图所示, U″′(916.1 eV)、V″′(897.6 eV)、U(900.3 eV)、V(881.9 eV)、U″(906.9 eV)和V″(888.5 eV)归属于Ce4+, U′(901.9 eV)、U0(898.6 eV)、V′(883.9 eV)和V0(880.6 eV)归属于Ce3+(Anandan et al., 2013).如表 2所示, Pt/CeO2和CeO2中Ce3+的含量分别为21.3%、17.1%.负载Pt之后, 载体表面的Ce3+显著提高.一般认为Ce3+的含量越高, 催化剂表面氧空位含量越高.该结果与H2-TPR和UV Raman的结果相一致, 进一步说明负载的Pt纳米颗粒与CeO2载体间存在的相互作用力可以活化CeO2的晶格氧, 降低其氧空位形成能.
 |
| 图 4 各催化剂Pt 4f、Al 2p、Ce 3d的XPS谱图 Fig. 4 The XPS patterns of Pt 4f, Al 2p, Ce 3d for various samples |
| 表 2 各个样品的化学性质参数 Table 2 Chemical parameters of various samples |
图 5为催化剂Pt/CeO2和Pt/Al2O3在不同温度下催化氧化甲苯的转化率.通常以T50和T90(甲苯转化率分别为50%和90%的反应温度)来衡量催化剂的催化活性.由图可知, Pt/CeO2催化剂的T50和T90分别为170和190 ℃, 而Pt/Al2O3的T50、T90分别为180和220 ℃.该结果表明, Pt/CeO2催化氧化甲苯效果明显高于Pt/Al2O3.在铂基催化剂催化氧化甲苯反应中, 暴露的Pt原子常被认为是反应活性位点, 起到吸附活化甲苯分子及解离气相氧的关键作用(Chen et al., 2013; Liotta, 2010).因此, 一般认为Pt的颗粒尺寸越小, 分散度越高, 材料所暴露的Pt活性位越多, 催化活性也就越好.本研究中, Pt/Al2O3的Pt分散度高于Pt/CeO2, 暴露出更多的Pt原子, 但催化活性却明显差于Pt/CeO2.接下来将利用TPD、GC/MS和In-situ FTIR技术, 从反应过程的角度做进一步探究.
 |
| 图 5 甲苯去除率随反应温度变化情况 Fig. 5 Conversion of toluene over Pt/CeO2 and Pt/Al2O3 catalysts |
图 6为甲苯TPD实验中各催化剂甲苯、H2、CO2的MS信号随温度的变化图.如图 6a所示, 随着温度升高, 所有样品均在50~70 ℃区间内出现了甲苯脱附峰, 其中, Pt/CeO2和Pt/Al2O3的甲苯脱附峰温度分别为68.8和66.4 ℃.甲苯在Pt/CeO2催化剂上的脱附温度较高, 这可能是因为甲苯与其结合更为紧密.而经过O2预处理过后的2个催化剂, 其甲苯脱附温度有所降低, 这可能是由于气相氧与甲苯在催化剂表面存在竞争吸附关系, 气相氧优先占据了部分吸附位点, 降低了甲苯的脱附温度(张薇薇, 2011).
 |
| 图 6 Pt/CeO2和Pt/Al2O3的甲苯TPD谱图(a.甲苯, b.H2, c.CO2) Fig. 6 TPD spectra of Pt/CeO2 and Pt/Al2O3 catalysts |
图 6b为甲苯TPD过程中H2的信号变化图.在甲苯脱附温度区间内, Pt/CeO2检测到H2的信号, 而Pt/Al2O3、O2-Pt/CeO2、O2-Pt/Al2O3均未检出.研究结果表明(Lai et al., 2014), 配位数更低的Pt原子具有更强的甲苯解离能力, 可以直接使苯基和甲基间的C—C链发生断裂, 并进一步分解为H2和苯.而在后续的反应副产物分析中, 如图 8所示, Pt/CeO2的反应尾气检测到了微量的苯, 而Pt/Al2O3并未检出.该结果表明, 甲苯在Pt/CeO2表面也可能发生苯基和甲基间的C—C链断裂现象, 随着温度升高, 进一步生成H2脱附.
 |
| 图 8 Pt/CeO2和Pt/Al2O3催化氧化甲苯尾气GC/MS分析谱图 Fig. 8 GC/MS measurement on the off-gas from toluene oxidation over Pt/CeO2 and Pt/Al2O3 catalysts |
图 6c为甲苯TPD过程中CO2的MS信号图.未经O2预吸附处理的样品均未检测到CO2的信号, 而O2-Pt/CeO2、O2-Pt/Al2O3催化剂在140 ℃附近出现了CO2的信号峰.这是由于O2预吸附处理后, O2-Pt/CeO2、O2-Pt/Al2O3样品表面吸附了部分氧, 随着温度升高, 吸附氧和甲苯发生反应生成CO2.值得注意的是, O2-Pt/CeO2的CO2峰强度更高, 说明有更多的氧参与到反应中.研究表明, 贵金属催化氧化反应体系中, 不同载体会影响反应的供氧路径(Markus et al., 2011).对于惰性载体, 供氧主要来源于气相氧在贵金属上的吸附解离; 对于活性载体, 如CeO2、ZrO2-Bi2O3等, 除了贵金属解离气相氧之外, 载体本身还可以提供活性氧物种, 并直接参与反应.因此, 在O2-Pt/CeO2催化剂产生更多CO2是由于CeO2载体能提供更多的活性氧物种参与反应.
3.4 Pt-CO原位FTIR实验为研究Pt/CeO2和Pt/Al2O3上Pt原子周边的电子环境, 本课题组进行了CO吸附的FTIR实验.图 7为CO吸附在两催化剂表面的FTIR谱图, 由图可知, 两种催化剂都在1990~2150 cm-1之间出现包峰, 该峰属于CO在Pt原子上的线式吸附(Shen et al., 2014; Jiang et al., 2015).其中, Pt/CeO2样品的CO吸附峰可分为3个峰(2095、2045和1994 cm-1), Pt/Al2O3可分为2个吸附峰(2046和2014 cm-1).一般认为, CO吸附在Pt原子上的红外峰位与Pt原子的配位数密切相关, 配位数越低, 峰位越小(Bazin et al., 2005).结合相关文献, 可以推断位于高波段的2095 cm-1峰归属于CO分子吸附在Pt的平台面等饱和配位处, 而在2040及2000 cm-1左右的峰归属CO分子吸附在位于台阶、边角等不饱和配位的Pt原子上.由表 1可知, 较Pt/Al2O3催化剂, 负载在CeO2上的Pt颗粒尺寸大, 位于平台面处的饱和配位的Pt原子会更多, 这也就解释了为何仅在Pt/CeO2催化剂上发现了位于2095 cm-1处的吸附峰.
 |
| 图 7 Pt/CeO2和Pt/Al2O3的CO吸附FTIR图谱 Fig. 7 FT-IR spectra of CO adsorbed on Pt/CeO2 and Pt/Al2O3 catalysts |
值得注意的是, Pt/CeO2样品在低波段处出现的CO吸附峰(1994 cm-1)的峰位较Pt/Al2O3(2014 cm-1)向低波段偏移了20 cm-1.一般认为, CO分子与Pt原子的结合是通过电子的配对形成的, CO的5σ轨道的电子向Pt的d空轨道进行供给, 同时, Pt原子填充了电子的d轨道也会向CO分子的2π*反键轨道回馈电子(Jiang et al., 2015).当CeO2等金属氧化物作为载体时, Pt与载体间会产生强相互作用, 促进电子及物质的转移, 一方面可能会弱化铈与氧之间的结合力; 另一方面从载体CeO2向Pt传递的电子会提升Pt原子的电子密度, 降低部分Pt原子的配位数, 进而导致CO的红外吸附峰向低波段偏移(Jiang et al., 2015).研究表明, 高电子密度的Pt原子具有较高的催化活性, 促进有机分子中C—C键的断裂, 提升反应催化活性(Lai et al., 2014).结合甲苯-TPD的实验结果, 可以推论由于Pt与CeO2间的强相互作用, 负载在载体CeO2表面的Pt原子具有更高的电子密度和更强的活化甲苯的能力, 因此, 其甲苯催化氧化性能更强.
3.5 副产物GC/MS分析图 8为Pt/CeO2和Pt/Al2O3催化氧化甲苯的转化率达50%(对应温度分别为170和180 ℃)时尾气的GC/MS分析谱图.经浓缩后的尾气中均检测到醛类、酯类物质, 具体物质成分如表 3所示. Pt/CeO2催化剂共检测到7种物质, 除了苯之外还含有5种醛类和1种酯类; 而Pt/Al2O3催化剂的尾气中都含有较多副产物, 共检测到10种物质, 包括1种醚类、4种醛类和5种酯类物质.结果表明, 在同等的甲苯转化率下, 相比于Pt/CeO2催化剂, Pt/Al2O3催化剂反应形成的副产物更多, 且较多为酯类.其分析结论将在下文结合红外结果分析.
| 表 3 Pt/CeO2和Pt/Al2O3催化氧化甲苯尾气GC/MS物质 Table 3 GC/MS species of the off-gas from toluene oxidation over Pt/CeO2 and Pt/Al2O3 catalysts |
图 9为Pt/CeO2催化剂在不同温度条件下催化氧化甲苯的原位红外谱图.由图可知, 通入甲苯混合气后, 在1495和1602 cm-1处出现了2个特征峰, 归属于甲苯的苯环骨架振动峰(Hernández-Alonso et al., 2011), 表明甲苯吸附在Pt/CeO2催化剂表面.随着温度升高, 苯环振动峰逐渐减弱, 说明Pt/CeO2催化剂表面的甲苯逐渐被降解.温度为50 ℃时, 在1303 cm-1处出现的特征峰归属于碳氢类物质的振动(Chen et al., 1996); 此外, 在1716、1734、1845、1868 cm-1处出现的特征峰分别归属于羰基、醛基的振动峰(Zhu et al., 2016; Fan et al., 2001; Bulsink et al., 2016), 说明甲苯可能被氧化为苯甲醛.当温度上升至150 ℃时, 代表醛酮类物质的峰减弱, 与此同时, 在1504、1548 cm-1处出现微弱的峰, 对应于苯酸盐和羧酸盐的振动峰(Viinikainen et al., 2013; Bao et al., 2011); 另外, 在1147、1176 cm-1处出现了v(C—O)的振动峰(Wang et al., 2012; Lochar et al., 2001).这些结果表明随着温度上升, 有更多的醛酮类物质被转化为苯酸盐物质.同时还可以看到, 在150 ℃出现了1356、1373、1393 cm-1等归属于碳酸氢盐vs(HCOO—)的振动峰(Araña et al., 2001; Innocent et al., 2010), 以及1589 cm-1处归属于水的羟基振动峰(Dos Santos et al., 2008).说明甲苯在催化剂表面被完全分解为H2O和CO2, 并在表面形成碳酸氢盐物质.当温度升高至190 ℃, 代表醛酮类物质的峰完全消失, 而碳酸氢盐物质峰明显增强.表明温度到达190 ℃时, 甲苯被完全氧化, 大量醛类、酸类产物被迅速分解为H2O和CO2.
 |
| 图 9 Pt/CeO2催化剂降解甲苯的原位红外分析(a.1100~1700 cm-1, b.1700~2000 cm-1) Fig. 9 In-situ FT-IR spectra of toluene degradation over Pt/CeO2 catalysts |
图 10为Pt/Al2O3催化剂在不同温度条件下催化氧化甲苯的原位红外谱图.由图中看出, 在室温下出现了1495和1602 cm-1峰, 表明甲苯吸附于Pt/Al2O3催化剂表面.随着温度升高, 该峰强度逐渐减弱, 表明催化剂表面上的甲苯正在被降解.在50 ℃时, 1716、1734 cm-1等醛酮类产物的振动峰出现, 说明甲苯可能进一步生成了苯甲醛; 温度上升到150 ℃时, 图中出现的1424、1508、1542、1598 cm-1特征峰分别代表长链羧酸盐和苯酸盐的振动峰(Driessen et al., 1998; Viinikainen et al., 2013; Bao et al., 2011; Dobson et al., 2000).此外, 在该温度下, 在1393 cm-1处出现了归属于碳酸氢盐的振动峰, 表明苯甲醛转化为苯酸盐类物质, 并且最终被完全分解为CO2.随着温度的升高, 代表醛酮类物质、苯酸盐类物质峰的强度也随之增强, 说明有部分醛酮类物质、苯酸盐类物质在表面累积, 并可能进一步反应为其他副产物.该结果与GC/MS检测结果相一致, Pt/Al2O3催化剂在T50时检测到多种醛类、酯类物质.
 |
| 图 10 Pt/Al2O3催化剂降解甲苯的原位红外分析(a.1100~1700 cm-1, b.1700~2000 cm-1) Fig. 10 In-situ FT-IR spectra of toluene degradation over Pt/Al2O3 catalysts |
对比两种催化剂红外及GC/MS结果发现, 在Pt/Al2O3催化剂表面存在醛、苯酸盐类物质累积的现象, 温度到达180 ℃时, 在反应尾气中依然会检测到较多醛和酯类物质.而在Pt/CeO2催化剂表面, 反应过程中生成的醛、苯酸盐类物质可以更快地反应为H2O和CO2.这可能是由于在Pt/CeO2催化剂表面存在的活性氧物种较Pt/Al2O3更为充足.对于Pt/Al2O3催化剂, 反应供氧主要来源于气相氧在Pt上的吸附解离, 供氧速率相对有限, 反应生成的中间物种来不及进一步氧化分解生成CO2和H2O, 而在催化剂表面累积并进一步相互反应形成其他副产物.对于Pt/CeO2催化剂, 由H2-TPR、Raman及甲苯TPD结果可知, Pt纳米颗粒的负载促进了CeO2氧空位形成, 进一步提高了CeO2储-放氧性能, 促进了氧循环(活性氧的释放与补偿), 能为甲苯的催化氧化提供更多的活性氧物种, 因此, 中间产物及副产物的累积相对较少.
结合本研究结果及相关文献结果可以推测, Pt/Al2O3的Pt分散度高于Pt/CeO2, 而催化氧化甲苯活性低的原因可能是由于甲苯在两催化剂上的反应机制不同.对于Pt/Al2O3催化剂, 甲苯在Pt颗粒上吸附并活化为脱氢中间产物, 气相氧在Pt颗粒上吸附解离, 再进一步反应为CO2和H2O.而对于Pt/CeO2催化剂, 除了上述反应路径外, 还可能存在以下机制:①由于Pt与CeO2间的强相互作用, 负载在载体CeO2表面的Pt原子具有更高的电子密度, 具有更强的活化甲苯能力, 可以直接使苯基和甲基间的C—C链发生断裂, 使甲苯分子更容易与活性氧反应; ②金属与载体间的强相互作用力活化了CeO2晶格氧, 提升了氧空位浓度, 加速氧循环, 为反应提供了更多的活性氧物种, 进一步提高甲苯催化氧化效率.
4 结论(Conclusions)1) 使用浸渍法成功制备Pt负载量约0.6%的Pt/CeO2和Pt/Al2O3催化剂, 其中, Al2O3载体上Pt颗粒尺寸小于Pt/CeO2, 而Pt/CeO2催化剂具有更好的甲苯催化氧化活性, 其T50和T90分别为170和190 ℃.
2) Pt/CeO2的Pt颗粒粒径大于Pt/Al2O3, 而催化活性更高是由于甲苯在两催化剂上的反应机制不同, Pt/CeO2催化剂催化氧化甲苯还存在以下机制:由于Pt与CeO2间的强相互作用, 负载在CeO2表面的Pt原子具有更高的电子密度, 使甲苯分子更容易进一步反应; CeO2载体可以提供活性氧物种, 直接参与催化氧化甲苯的反应, 提高甲苯催化氧化效率.
Agarwal S, Zhu X, Hensen E J M, et al. 2014. Defect chemistry of ceria nanorods[J]. The Journal of Physical Chemistry C, 118: 4131–4142.
DOI:10.1021/jp409989y
|
Anandan C, Bera P. 2013. XPS studies on the interaction of CeO2 with silicon in magnetron sputtered CeO2 thin films on Si and Si3N4 substrates[J]. Applied Surface Science, 283: 297–303.
DOI:10.1016/j.apsusc.2013.06.104
|
Araña J, González Díaz O, Miranda Saracho M, et al. 2001. Photocatalytic degradation of formic acid using Fe/TiO2 catalysts:the role of Fe3+/Fe2+ ions in the degradation mechanism[J]. Applied Catalysis B:Environmental, 32: 49–61.
DOI:10.1016/S0926-3373(00)00289-7
|
Bao Y, Ma J, Li N. 2011. Synthesis and swelling behaviors of sodium carboxymethyl cellulose-g-poly(AA-co-AM-co-AMPS)/MMT superabsorbent hydrogel[J]. Carbohydrate Polymers, 84(1): 76–82.
DOI:10.1016/j.carbpol.2010.10.061
|
Bazin P, Saur O, Lavalley J C, et al. 2013. FT-IR study of CO adsorption on Pt/CeO2:characterisation and structural rearrangement of small Pt particles[J]. Physical Chemistry Chemical Physics, 7(1): 187–194.
|
Borgna A F, Normand Le, Garetto T, et al. 1992. Sintering of Pt/A12O3 reforming catalysts:EXAFS study of the behavior of metal particles under oxidizing atmosphere[J]. Catalysis Letters, 13: 175–188.
DOI:10.1007/BF00770989
|
Bulsink P, Al-Ghamdi A, Joshi P, et al. 2016. Capturing Re (Ⅰ) in a neutral N, N, N pincer Scaffold and resulting enhanced absorption of visible light[J]. Dalton Transactions, 45(21): 8885–8896.
DOI:10.1039/C6DT00661B
|
Calaza F C, Xu Y, Mullins D R, et al. 2012. Oxygen vacancy-assisted coupling and enolization of acetaldehyde on CeO2 (111)[J]. Journal of the American Chemical Society, 134(43): 18034–18045.
DOI:10.1021/ja3074243
|
Chang S, Li M, Hua Q, et al. 2012. Shape-dependent interplay between oxygen vacancies and Ag-CeO2 interactionin Ag/CeO2 catalysts and their influence on the catalytic activity[J]. Journal of Catalysis, 293: 195–204.
DOI:10.1016/j.jcat.2012.06.025
|
Chen C Y, Zhu J, Chen F, et al. 2013. Enhanced performance in catalytic combustion of toluene over mesoporous Beta zeolite-supported platinum catalyst[J]. Applied Catalysis B:Environmental, 140-141: 199–205.
DOI:10.1016/j.apcatb.2013.03.050
|
Chen L, Lin L, Xu Z, et al. 1996. Fourier transform-infrared investigation of adsorption of methane and carbon monoxide on HZSM-5 and Mo/HZSM-5 zeolites at low temperature[J]. Journal of Catalysis, 161: 107–114.
DOI:10.1006/jcat.1996.0167
|
Dobson K D, McQuillan A J. 2000. In situ infrared spectroscopic analysis of the adsorption of aromatic carboxylic acids to TiO2, ZrO2, Al2O3, and Ta2O5 from aqueous solutions[J]. Spectrochimica Acta Part A, 56: 557–565.
DOI:10.1016/S1386-1425(99)00154-7
|
Dos Santos M L, Lima R C, Riccardi C S, et al. 2008. Preparation and characterization of ceria nanospheres by microwave-hydrothermal method[J]. Materials Letters, 62(30): 4509–4511.
DOI:10.1016/j.matlet.2008.08.011
|
Driessen M D, Miller T M, Grassian V H. 1998. Photocatalytic oxidation of trichloroethylene on zinc oxide:characterization of surface-bound and gas-phase products and intermediates with FT-IR spectroscopy[J]. Journal of Molecular Catalysis A:Chemical, 131: 149–156.
DOI:10.1016/S1381-1169(97)00262-8
|
Fan Q G, Lewis D M, Tapley K N. 2001. Characterization of cellulose aldehyde using Fourier transform infrared spectroscopy[J]. Journal of Applied Polymer Science, 82(5): 1195–1202.
DOI:10.1002/(ISSN)1097-4628
|
Gao Y, Wang W, Chang S, et al. 2013. Morphology effect of CeO2 support in the preparation, metal-support interaction, and catalytic performance of Pt/CeO2 catalysts[J]. Chem Cat Chem, 5(12): 3610–3620.
|
Genuino H C, Dharmarathna S, Njagi E C, et al. 2012. Gas-phase total oxidation of benzene, toluene, ethylbenzene, and xylenes using shape-selective manganese oxide and copper manganese oxide catalysts[J]. The Journal of Physical Chemistry C, 116(22): 12066–12078.
DOI:10.1021/jp301342f
|
Hernández-Alonso M D, Tejedor-Tejedor I, Coronado J M, et al. 2011. Operando FTIR study of the photocatalytic oxidation of methylcyclohexane and toluene in air over TiO2-ZrO2 thin films:Influence of the aromaticity of the target molecule on deactivation[J]. Applied Catalysis B:Environmental, 101(3/4): 283–293.
|
Huang H, Dai Q, Wang X. 2014. Morphology effect of Ru/CeO2 catalysts for the catalytic combustion of chlorobenzene[J]. Applied Catalysis B:Environmental, 158-159: 96–105.
DOI:10.1016/j.apcatb.2014.01.062
|
Innocent B, Pasquier D, Ropital F, et al. 2010. FTIR spectroscopy study of the reduction of carbon dioxide on lead electrode in aqueous medium[J]. Applied Catalysis B:Environmental, 94(3/4): 219–224.
|
Jang F, Zeng L, Liu G, et al. 2015. Propane dehydrogenation over Pt/TiO2-Al2O3 catalysts[J]. ACS Catalysis, 5: 438–447.
DOI:10.1021/cs501279v
|
Lai Y T, Chen T C, Lan Y K, et al. 2016. Pt/SBA-15 as a highly efficient catalyst for catalytic toluene oxidation[J]. ACS Catalysis, 4: 3824–3836.
|
Lee S, Seo J, Jung W. 2016. Sintering-resistant Pt@CeO2 nanoparticles for high-temperature oxidation catalysis[J]. Nanoscale, 8(19): 10219–10228.
DOI:10.1039/C6NR00170J
|
廖银念. 2013. 铈基金属氧化物催化氧化甲苯的形貌及尺寸效应[D]. 广州: 华南理工大学. 128
http://cdmd.cnki.com.cn/Article/CDMD-10561-1014153382.htm |
Liotta L F. 2010. Catalytic oxidation of volatile organic compounds on supported noble metals[J]. Applied Catalysis B:Environmental, 100: 403–412.
DOI:10.1016/j.apcatb.2010.08.023
|
Liu H H, Wang Y, Jia A P, et al. 2014. Oxygen vacancy promoted CO oxidation over Pt/CeO2 catalysts:A reaction at Pt-CeO2 interface[J]. Applied Surface Science, 314: 725–734.
DOI:10.1016/j.apsusc.2014.06.196
|
Lochar V, Machek J, Tichy J. 2002. Mechanism of selective oxidation of methanol over stannic oxide-molybdenum oxide catalyst[J]. Applied Catalysis A:General, 228: 95–101.
DOI:10.1016/S0926-860X(01)00970-X
|
López J M, Gilbank A L, García T, et al. 2015. The prevalence of surface oxygen vacancies over the mobility of bulk oxygen in nanostructured ceria for the total toluene oxidation[J]. Applied Catalysis B:Environmental, 174-175: 403–412.
DOI:10.1016/j.apcatb.2015.03.017
|
Markus M S, Stefan H, Andre C V, et al. 2001. CO Oxidation over supported gold catalysts-"inert" and "active" support materials and their role for the oxygen supply during reaction[J]. Journal of Catalysis, 197: 113–122.
DOI:10.1006/jcat.2000.3069
|
Mcbride J R, Hass K C, Poindexter B D, et al. 1994. Raman and x-ray studies of Ce1-xRExO2-y, where RE=La, Pr, Nd, Eu, Gd, and Tb[J]. Journal of Applied Physics, 76(4): 2435.
DOI:10.1063/1.357593
|
Moreno-González M, Blasco T, Góra-Marek K, et al. 2014. Study of propane oxidation on Cu-zeolite catalysts by in-situ EPR and IR spectroscopies[J]. Catalysis Today, 227: 123–129.
DOI:10.1016/j.cattod.2013.10.055
|
Peng R, Sun X, Li S, et al. 2016. Shape effect of Pt/CeO2 catalysts on the catalytic oxidation of toluene[J]. Chemical Engineering Journal, 306: 1234–1246.
DOI:10.1016/j.cej.2016.08.056
|
Reyes P, Pecchi G, Morales M, et al. 1997. The nature of the support and the metal precursor on the resistance to sulphur poisoning of Pt supported catalysts[J]. Applied Catalysis A:General, 163(1997): 145–152.
|
Rui S, Maria F S. 2008. Shape and crystal-plane effects of nanoscale ceria on the activity of Au-CeO2 catalysts for the water-gas shift reaction[J]. Angewandte Chemie International Edition, 120: 2926–2929.
|
Ruppert A M, Paryjczak T. 2007. Pt/ZrO2/TiO2 catalysts for selective hydrogenation of crotonaldehyde:Tuning the SMSI effect for optimum performance[J]. Applied Catalysis a:General, 320: 80–90.
DOI:10.1016/j.apcata.2006.12.019
|
Shen M Q, Lv L F, Wang J Q, et al. 2014. Study of Pt dispersion on Ce based supports and the influence on the CO oxidation reaction[J]. Chemical Engineering Journal, 306: 1234–1246.
|
Vayssilov G N, Lykhach Y, Migani A, et al. 2011. Support nanostructure boosts oxygen transfer to catalytically active platinum nanoparticles[J]. Nature materials, 10(4): 310–315.
DOI:10.1038/nmat2976
|
Viinikainen T, Kauppi I, Korhonen S, et al. 2013. Molecular level insights to the interaction of toluene with ZrO2-based biomass gasification gas clean-up catalysts[J]. Applied Catalysis B:Environmental, 142-143: 769–779.
DOI:10.1016/j.apcatb.2013.06.008
|
Wang C, Feng L, Li W, et al. 2012. Shape-stabilized phase change materials based on polyethylene glycol/porous carbon composite:The influence of the pore structure of the carbon materials[J]. Solar Energy Materials and Solar Cells, 105: 21–26.
DOI:10.1016/j.solmat.2012.05.031
|
Wang T, Jiang F, Liu G, et al. 2016. Effects of Ga doping on Pt/CeO2-Al2O3 catalysts for propane dehydrogenation[J]. AIChE Journal, 62(12): 4365–4376.
DOI:10.1002/aic.15339
|
Weber W H, Hass K C, Mcbride J R. 1993. Raman study of CeO2:second-order scattering, lattice dynamics, and particle-size effects[J]. Physical Review B, 48(1): 178–185.
DOI:10.1103/PhysRevB.48.178
|
Wu Z L, Li M j, Howe J, et al. 2016. Probing defect sites on CeO2 nanocrystals with well-defined surface planes by raman spectroscopy and O2 adsorption[J]. Langmuir, 26(21): 1659–16606.
|
Zhang C, He H, Tanaka K. 2006. Catalytic performance and mechanism of a Pt/TiO2 catalyst for the oxidation of formaldehyde at room temperature[J]. Applied Catalysis B:Environmental, 65(1/2): 37–43.
|
Zhu X, Sun J, Wang S, et al. 2016. Synthesis, crystal structure, superoxide scavenging activity, anticancer and docking studies of novel adamantyl nitroxide derivatives[J]. Journal of Molecular Structure, 1108: 611–617.
DOI:10.1016/j.molstruc.2015.12.048
|
张薇薇. 2011. 甲苯在SBA-15上吸附/催化性能的研究[D]. 大连: 大连理工大学. 46
http://cdmd.cnki.com.cn/Article/CDMD-10141-1011108484.htm |