 2017, Vol. 37
2017, Vol. 37



2. 燃料电池湖北省重点实验室, 武汉 430070
2. Key Laboratory of Hubei Province for Fuel Cell, Wuhan 430070
厌氧消化技术目前广泛用于污泥及高浓度有机废水的处理并可回收沼气, 但厌氧消化液中存在高浓度氨氮及硫化物.研究发现, 某些工业废水如制革废水和采矿废水等也会产生高浓度的硫化物及氮素污染物(Guo et al., 2016).硫化物具有臭味和腐蚀性, 会严重影响人体健康及生活环境, 氮素污染物则是水体富营养化的主要诱因, 因此, 这些废水排放前需除硫脱氮.近年来出现的微生物燃料电池(Microbial Fuel Cell, MFC)可在去除污染物的同时回收电能, 在废水脱氮或废水除硫领域具有较好的发展前景(Sun et al., 2016; Zhao et al., 2008).前期的MFC除硫多采用化学阴极, 以高锰酸钾或铁氰化钾为电子受体(Cai et al., 2015; Lee et al., 2012), 易产生二次污染.以S2-作为阳极电子供体, NO3-为阴极电子受体, 可在单一的反硝化除硫MFC内分别完成阳极除硫与阴极脱氮(魏炎等, 2016), 具有处理含S2-/NH4+废水的潜力.
值得注意的是, 废水中的氮主要以NH4+形式存在, 采用反硝化除硫MFC处理含S2-/NH4+废水之前, 需要先将NH4+氧化为NO3-.主要方法有:在阳极和阴极之间外接硝化反应器产生NO3-(Virdis et al., 2008); 耦合好氧生物阴极MFC和反硝化MFC, 以好氧阴极MFC产生的NO3-为反硝化MFC提供阴极电子受体(Xie et al., 2011); 直接将含NH4+废水充氧, 在MFC阴极进行同步硝化/反硝化(Virdis et al.2010).相比而言, 在MFC阴极进行同步硝化/反硝化时, 氧作为更强的电子受体会抑制反硝化效果, 需谨慎控制阴极曝气量.因此, 如以MFC同时进行阳极除硫与阴极硝化, 可克服化学阴极的缺点, 阴极硝化产生的NO3-还能为将来的反硝化除硫MFC提供阴极电子受体, 降低了氧对阴极反硝化的影响, 但这方面的研究目前还鲜见相关报道.
MFC除硫时, S2-在阳极被氧化为S和SO42-, 阳极微生物作用加速了S向SO42-的转换(Sun et al., 2009).含硫废水处理中产生的SO42-直接排入水体时, 易于在厌氧条件下重新生成S2-, 造成水体的二次污染.如定期清洗并更换阳极电极, 通过清除阳极微生物的方式减少SO42-的形成, 既有利于回收硫资源, 又能降低硫的二次污染.换水周期长短也可影响S2-氧化的产物形式, 因此, 有必要研究换水周期内的S2-转化情况.
本研究采用MFC处理含S2-/NH4+废水同时回收电能, 含S2-/NH4+废水先在阳极去除S2-, 阳极出水再进入阴极完成NH4+氧化.同时, 研究不同进水S2-浓度下的除硫硝化及产电性能, 并探讨不同阳极碳刷清洗周期和换水周期内的污染物去除、产电性能及阳极室硫累积情况, 为MFC处理含S2-/NH4+废水奠定基础.
2 材料与方法(Materials and methods) 2.1 除硫硝化MFC双室型除硫硝化MFC采用有机玻璃制成(图 1), 阴、阳极室以经预处理(于景荣等, 2001)后的质子膜(Dupont, 美国)隔开.MFC的阴、阳极室的有效容积均为300 cm3, 以碳刷(新浩特, 荆州)作为电极.阴、阳极通过可调电阻箱连接成回路, 外电阻设为100 Ω.阴极室通过气泵曝气供氧, 阳极室通过底部的磁力搅拌器混合阳极液.除硫硝化MFC采用分批进水方式运行, 每隔11.5 h更换进水(即换水周期为11.5 h).环境温度为(25.0±0.5) ℃.
 |
| 图 1 除硫硝化MFC装置示意图 Fig. 1 Schematic of sulfide removal and nitrification MFC |
阳极进水为含S2-/NH4+的模拟无机废水, 其基础营养溶液成分为:NaHCO3 8.4 g·L-1, MgCl2 4.25 mg·L-1, CaCl2 2.5 mg·L-1, 微量元素溶液1 mL·L-1(Virdis et al., 2008).另以Na2S提供阳极电子供体, NH4Cl(28 mg·L-1)作为氮源.阴极进水在第1个换水周期时的主要成分为上述基础营养溶液(其中, NaHCO3浓度改为0.5 g·L-1)加NH4Cl(28 mg·L-1), 随后的换水周期以阳极出水为阴极进水.阳极不接种污泥, 阴极接种污泥取自武汉市龙王嘴污水处理厂缺氧池.
2.3 研究方案除硫硝化MFC前期已在试验室完成启动.将阳极进水S2-浓度分别设定为(60.8±2.9)、(131.7±2.4)、(161.7±4.5) 和(198.1±3.1) mg·L-1进行本试验.为抑制阳极微生物作用, 促进S的生成, 定期清洗并更换阳极碳刷.阳极出水SO42-浓度发生骤升前更换并清洗阳极碳刷, 即为一个碳刷清洗周期.每个浓度下进行2个碳刷清洗周期试验.第1个碳刷清洗周期内, 分析各换水周期的污染物去除和产电情况, 并测定极化曲线.第2个碳刷清洗周期内, 分析最后一个换水周期中不同时间段的污染物去除及产电情况, 最后取出阳极碳刷和阳极液测定碳刷清洗周期内的沉积硫累积量.对应每个进水S2-浓度设置3组无菌对照试验, 对照1的阴、阳极开路且阴极不曝气, 对照2的阴、阳极闭路但阴极不曝气, 对照3的阴、阳极闭路且阴极曝气.每组对照重复3个换水周期, 探究除硫硝化MFC的两极反应途径.对照试验前, MFC装置采用紫外灭菌灯照射20 min, 模拟废水的基础溶液先通过121 ℃灭菌处理, 再以0.22 μm滤头过滤加入相应的S2-和NH4+, 进水方式与前述试验相同.
2.4 测试与分析方法S2-和NH4+-N采用标准方法分析(国家环境保护局, 2002), SO42--S、NO3--N和NO2--N采用ICS-900型离子色谱仪(Dionex, 美国)测定.沉积硫参照文献方法测定(Jiang et al., 2009).采用UT71D万用电表每4 min记录一次输出电压U, 根据欧姆定律计算相应的电流I及功率密度P.通过稳态放电法拟合MFC极化曲线(梁鹏等, 2007).阳极库仑效率CE采用式(1) 计算, 实际电荷量CA采用式(2) 计算, 理论释放电荷量CT采用式(3) 计算.
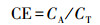
|
(1) |
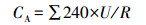
|
(2) |
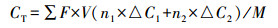
|
(3) |
式中, 240为记录时间间隔(s), U为电压(V), R为外阻值(100 Ω), F为法拉第常数(96485 C·mol-1), V为阳极或阴极周期换水量(0.3 L), n1=2, △C1为阳极进出水S2-浓度差(g·L-1), n2=6, △C2为阳极进出水SO42-浓度差(g·L-1), M为硫的摩尔质量(32 g·mol-1).
采用ULTRA plus型电子显微镜(Zeiss, 德国)观察阳极碳纤维丝沉积物形态.(131.7±2.4) mg·L-1 S2-浓度下第1个清洗周期结束后, 离心分离阳极碳纤维丝及悬浮液沉积物, 冷冻干燥后采用ESCALAB 250Xi型X-射线光电子能谱(XPS) (Thermo Fisher, 美国)分析S2-氧化后的价态分布.以非单色化的Mg Kα X-射线(1253.6 eV)作为激发源, 分析室底真空度低于1×10-8 Pa, 以石墨C1s结合能284.3 eV作为标准对其它谱线校正.分峰拟合时, 采用Shirley本底扣除法对包络峰扣除本底, Gauss-Lorentz混合型函数对谱峰解叠化.
3 结果与讨论(Results and discussion) 3.1 除硫硝化MFC的反应机理分析如表 1所示, 对照3(即无菌对照MFC)中产生了电量, 表明除硫硝化MFC可通过非生物电化学作用产电.相同进水浓度下对照1、2、3的阳极S2-去除率依次增加, 对照1中未检测到SO42-, 而对照2和3中形成了少量SO42-(一般约2.2~8.2 mg·L-1), 这是由于不同对照试验中发生的阳极反应不同.好氧阴极MFC中, 氧能透过质子膜从阴极室向阳极室渗漏(Chae et al., 2008), 进而对阳极S2-的氧化去除产生影响.对照1只能存在S2-的自发化学氧化, 对照2存在S2-的自发化学氧化和好氧化学氧化, 对照3则存在S2-的自发化学氧化、好氧化学氧化和非生物电化学氧化.正常运行的除硫硝化MFC中还存在微生物作用.不同对照中的SO42-累积量不同则表明自发化学氧化不会形成SO42-, 而好氧氧化和自发电化学作用可形成少量SO42-.进水S2-浓度为(61.5±2.3) mg·L-1时, 对照3的SO42-累积高达(15.4±3.0) mg·L-1, 则是由于阳极S2-几乎耗尽( < 3.0 mg·L-1), 阴极非生物电化学作用仍需从阳极索取电子, 推动了硫向高价态氧化.Sun等(2009)得出了与此不同的结论:非生物电化学作用难于将S2-氧化为SO42-, 将S2-氧化为SO42-需要微生物作用完成.其可能原因在于:Sun等(2009)采用了表面积较小的阳极材料(亲水碳纸), S2-氧化为S和低价硫化合物能够为阳极提供充足的电子, 非生物电化学作用将S和低价硫化合物进一步氧化为SO42-的推动力不足.
| 表 1 无菌对照试验的S2-转化及产电量 Table 1 Sulfide transformation and charge recovery in control MFC |
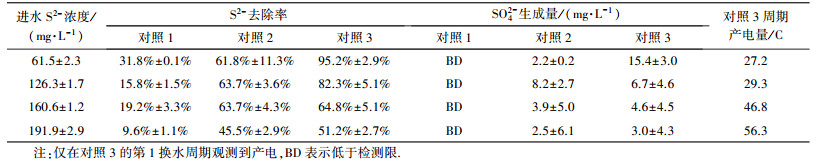
无菌对照试验中未发现阴极硝化作用(数据未列出), 但除硫硝化MFC的阴极硝化完全, 说明NH4+的氧化只能通过硝化菌完成.据报道(Freguia et al., 2010; You et al., 2009), MFC好氧生物阴极中可同时存在硝化菌和好氧电活性微生物, 硝化菌不参与电化学反应, 但电活性微生物能以氧为电子受体氧化由阳极传递来的电子, 进而提高MFC的产电能力.在前期完成的除硫硝化MFC启动过程中, 观察到周期产电量从初期的20 C逐步上升到60 C, 则是电活性微生物逐步增殖并提高产电能力的反映.
比较发现, 相近进水S2-浓度下表 2中的产电量明显高于表 1中对照3的产电量, 这是因为无菌对照条件下MFC只通过非生物电化学作用产电(表 1), 而正常运行的MFC通过非生物电化学作用和生物电化学作用共同产电(表 2).相近进水S2-浓度下, 对照3产电量(表 1)与MFC产电量(表 2)的比值可近似看作非生物电化学产电量占MFC产电量的比例.根据第1换水周期的产电量, 计算出非生物电化学产电量占MFC产电量的34.9%±7.1%.
| 表 2 不同进水S2-浓度下MFC的污染物去除 Table 2 Pollutants removal of MFC at different sulfide feeding concentrations |
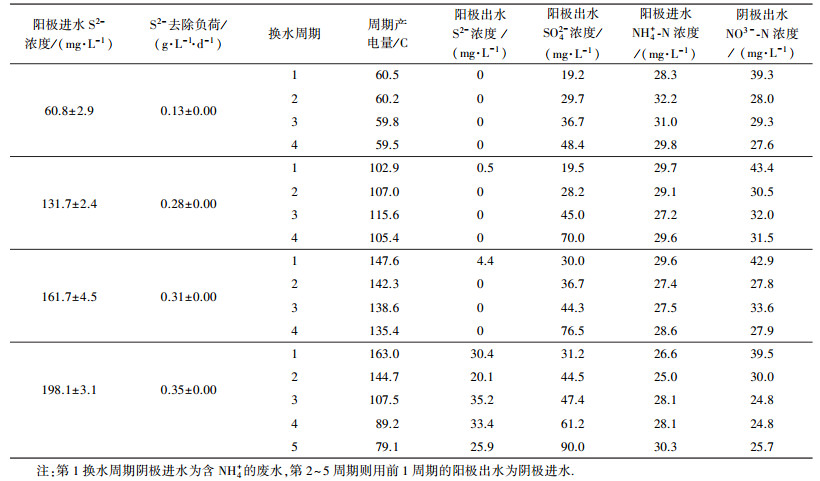
如表 2所示, S2-去除负荷随进水S2-浓度的增加而增加, 但进水S2-浓度为(161.7±4.5) mg·L-1时第1周期出水有少量S2-残余(4.4 mg·L-1), 进水S2-浓度为(198.1±3.1) mg·L-1时各周期出水均有较多的S2-残余(20.1~35.2 mg·L-1), 表明进水S2-浓度不宜超过161.7 mg·L-1.阳极出水SO42-浓度随换水周期数及进水浓度的增加而增加.进水S2-浓度为(60.8±2.9)、(131.7±2.4)、(161.7±4.5)、(198.1±3.1) mg·L-1时, 根据SO42-骤升确定相应的碳刷清洗周期分别为3、3、3和4个换水周期.本研究中阳极未接种污泥, 但阳极进水也未灭菌处理, 因此, MFC长期运行过程中仍可能引入硫氧化菌, 并在阳极室器壁及质子膜上繁殖.与无菌对照试验相比(表 1), 相近进水浓度下MFC的S2-去除率和SO42-产量更高.其主要原因在于:① 阳极室器壁及质子膜上硫氧化菌的生物氧化作用加速了S2-的去除和SO42-的生成; ② 阴极电活性微生物的电化学作用推动了阳极S2-的去除和SO42-的生成.
改变阳极S2-进水浓度对阴极硝化效果无影响, 各浓度下阴极出水NH4+-N及NO2--N的浓度低于检测限, 硝化完全(数据未列出).这是因为由阳极传递到阴极的电子主要通过生物和非生物电化学作用被氧化(而不是被硝化菌利用), 硝化菌不参与阴极电化学反应, 其硝化效果主要与供氧量有关.试验发现, 换水周期结束时MFC阳极室存在显著的NH4+损失(损失率约35.2%~58.0%).由于阳极室没有接种污泥, 且进水pH维持在中性, 推测NH4+由阳极室透过质子膜向阴极室发生了迁移并被氧化为NO3-. Kim等(2008)在MFC研究中也发现了NH4+由阳极室向阴极室的迁移.
3.2.2 阳极碳刷清洗周期内MFC的产电性能如图 2a所示, 最大功率密度随着阳极进水S2-浓度的增加而提升, 阳极进水S2-浓度为(60.8±2.9)、(131.7±2.4) 和(161.7±4.5) mg·L-1时, 最大功率密度分别为3.39、4.15和5.77 W·m-3, 相应的周期产电量分别为(60.0±0.4)、(107.7±5.1) 和(141.0±5.2) C(根据表 2统计). Zhao等(2008)以MFC处理含SO42-废水时也发现, MFC电流一定程度上取决于S2-浓度的高低.当阳极进水S2-浓度从(161.7±4.5) mg·L-1上升到(198.1±3.1) mg·L-1时, 最大功率密度仅增加0.57 W·m-3.如图 2b所示, 进水S2-浓度由(60.8±2.9) mg·L-1提高至(131.7±2.4) mg·L-1时, 阳极库仑效率由21.5%±3.3%增加到32.9%±4.3%.但当进水S2-浓度进一步提高到(161.7±4.5) 和(198.1±3.1) mg·L-1时, 阳极库仑效率反而下降.可能原因是:阳极碳刷上的电子接受点位和阴极电活性微生物的电子消耗能力有限, 更多比例的S2-主要通过化学氧化作用及生物氧化作用得以去除, 生物氧化由阳极室器壁和质子膜上的硫氧化菌催化完成.在MFC阳极同时去除S2-与NO3-时也发现了高浓度S2-条件下阳极库仑效率低的现象(Cai et al., 2013).
 |
| 图 2 不同进水S2-浓度下MFC的功率密度曲线(a)和阳极库仑效率(b) Fig. 2 Power density (a) and anodic columbic efficiencies (b) at different sulfide feeding concentrations |
相同进水S2-浓度下, 同一洗涤周期内阳极库仑效率随换水周期的增加而减少.这是因为随着换水周期增加, 碳刷上的沉积硫增加, 阻碍了电化学反应的进行(Sun et al., 2009).进水S2-浓度为(198.1±3.1) mg·L-1时, 产电量由第1周期的163.0 C逐步下降到第5周期的79.1 C(表 2), 阳极库仑效率也随运行周期数的增加快速下降(图 2b).阳极出水作为下一周期阴极进水时, 残余的S2-可能对阴极电活性微生物产生抑制作用(Lee et al., 2012), 表明进水S2-浓度不宜超过(161.7±4.5) mg·L-1.本研究中阳极库仑效率总体较低( < 40%), 原因在于:氧透过质子膜从阴极室向阳极室渗漏(Chae et al., 2008), 进而引起S2-的化学氧化.后期研究中需合理控制阴极曝气量, 或外接硝化反应器完成硝化作用(Virdis et al., 2008).综合污染物去除及产电性能, 进水S2-浓度设为(161.7±4.5) mg·L-1较为适宜, 相应的S2-去除负荷为(0.31±0.00) g·L-1·d-1, 周期产电量为(141.0±5.2) C.
3.3 不同进水S2-浓度下换水周期内的MFC性能 3.3.1 换水周期内MFC的污染物去除性能如图 3所示, 进水S2-浓度为(60.8±2.9)、(131.7±2.4) 和(161.7±4.5) mg·L-1条件下的第3、3和3个换水周期内, S2-耗尽( < 0.5 mg·L-1)的时间依次为6、8和8 h, 与此同时阴极NH4+和NO2-均低于检测限, 硝化完全(数据未列出).但进水S2-浓度增加至(198.1±3.1) mg·L-1时, 11.5 h后仍有S2-残余(20.8 mg·L-1).Cai等(2013)也发现阳极S2-在5 h内从61.5 mg·L-1降至1.0 mg·L-1.如3.1节所述, 好氧化学氧化、非生物电化学反应及微生物反应共同催化了SO42-的生成.每个进水浓度下换水2 h内即有SO42-生成(4.0~18.5 mg·L-1), 表明MFC阳极难于使S2-全部氧化为S.每个进水浓度下SO42-浓度随运行时间缓慢增加, 待S2-耗尽后, 生成的S仍继续氧化生成SO42-, 硫素平衡分析可知S2-耗尽时S生成量接近最大值.
 |
| 图 3 不同进水S2-浓度下最后一个换水周期内的S2-和SO42-浓度变化(a.(60.8±2.9) mg·L-1条件下的第3个周期, b.(131.7±2.4) mg·L-1条件下的第3个周期, c.(161.7±4.5) mg·L-1条件下的第3个周期, d.(198.1±3.1) mg·L-1条件下的第4个周期) Fig. 3 Concentration variation of S2- and SO42-during the last batch at different sulfide feeding concentrations (a. the third batch for (60.8±2.9) mg·L-1, b. the third batch for (131.7±2.4) mg·L-1, c. the third batch for (161.7±4.5) mg·L-1, d. the fourth batch for (198.1±3.1) mg·L-1) |
如图 4所示, 各进水浓度下的MFC输出电压和阳极电势与阳极S2-浓度密切相关.结合图 3与图 4发现, 阳极S2-浓度低于6.2~17.4 mg·L-1时, 阳极电势上升至-23~-70 mV, 电压值开始快速下降.阳极S2-耗尽( < 0.5 mg·L-1)时, 阳极电势变为正值, 但电压并未降至0 mV(16~31 mV), S通过电化学作用进一步氧化成SO42-是MFC继续产电的原因.由图 4a可知, 每个进水S2-浓度下最后1个运行周期内MFC的输出电压总体呈先上升后下降的趋势, 更换废水后MFC需重新适应相应浓度的水质, 因此, 初始电压并非最高值, 后期随着S2-的消耗则电压逐步下降.进水S2-浓度为(198.1±3.1) mg·L-1时, 后期未出现电压下降, 则是由于11.5 h后仍有充足的电子供体S2- (20.8 mg·L-1).以硫化物和葡萄糖为阳极基质的MFC分批试验中存在类似的电压变化趋势(Zhang et al., 2009).增加进水浓度有利于MFC电压升高并延长稳定产电时间.进水S2-浓度为(60.8±2.9)、(137.1±2.4) 和(161.7±4.5) mg·L-1时, 换水周期内稳定电压的持续时间分别为4、6和6 h, 相应的最高电压分别为371、447和496 mV.进水S2-浓度升为(198.1±3.1) mg·L-1时, 11.5 h后仍可稳定产电( > 377 mV), 但上个换水周期阳极出水残余的S2-进入阴极后会抑制阴极反应, 导致最高电压回降到419 mV.综合MFC的产电与污染物去除情况, 进水S2-浓度为(60.8±2.9)、(137.1±2.4) 和(161.7±4.5) mg·L-1时的换水周期时间可分别缩短为6、8和8 h.
 |
| 图 4 不同进水S2-浓度下最后1个换水周期内的电压(a)和阳极电势(b)变化 Fig. 4 Voltage variation(a) and anode potential(b) during the last batch at different S2-concentrations |
SEM观察发现(图 5), 空白碳纤维丝表面较光滑, 阳极碳纤维丝上聚积有颗粒状物质, 结构较松散, 颗粒物呈圆形, 粒径较均匀, 大小约100 nm, 推测为颗粒硫.与其他研究者(远野等, 2014)观察到的的生物硫粒形态相似, 但颗粒更细.图 5b中还观测到阳极碳纤维丝上沉积物较厚时, 沉积物有明显的脱落现象, 这意味着碳纤维丝作为阳极电极材料将有利于后期的沉积硫清除与回收.
 |
| 图 5 阳极碳纤维丝FESEM图(a.空白碳纤维丝, ×1000; b.阳极碳纤维丝, ×1000; c.阳极碳纤维丝, ×10000;d.阳极碳纤维丝, ×50000) Fig. 5 FESEM images of anodic carbon fiber under different magnification (a.control carbon, ×1000;b.anodic carbon fiber, ×1000; c. anodic carbon fiber, ×10000; d.anodic carbon fiber, ×50000) |
XPS是重要的元素分析技术, 可用于分析硫在化合物中的状态及结合方式.如图 6所示, MFC阳极沉积物和悬浮物的XPS S2p谱图上有2个特征峰, 这2个峰的归属通过与文献谱图比较后而得到确认(https://srdata.nist.gov/xps/).159~166 eV处的宽峰通过解叠化分成一个双峰和一个单峰:在161.5 eV和162.6 eV处的S2p3/2-1/2双峰属于S2-, 而163.2 eV处的S2p3/2单峰被归于S0, 168.2 eV处的S2p3/2宽峰则是S6+的特征峰.阳极悬浮物XPS各个峰S6+:S0:S2-的相对峰面积比为1:0.42:0.31, 阳极沉积物的S6+:S0:S2-的相对峰面积比为1:0.11:0.06.悬浮物与沉积物相比, 悬浮物中S0含量比例较高, 而沉积物中S6+比例较高.可能是硫化物及硫沉积在碳纤维丝后易于进一步氧化, 因此, 如何控制碳纤维丝上的硫进一步氧化是回收硫的关键.Sun等(2009)采用以碳纸为阳极的MFC处理含S2-废水的研究发现, 在进水S浓度为133 mg·L-1条件下, 48 h时非生物MFC阳极沉积物中S6+:S0:S2-的相对峰面积比为1:0.93:0.46;随着时间延长, S0比例增加而S2-比例减小.相比本试验采用的碳刷阳极, Sun等(2009)采用的碳纸阳极具有较小表面积和较少的电化学作用位点, 使S0和S2-氧化为S6+的电化学推动力低于本研究, 因此, 其S0和S2-比例高于本试验结果.
 |
| 图 6 阳极悬浮物(a)和碳刷沉积物(b)的XPS能谱 Fig. 6 XPS energy spectrum for suspended solids in the anode chamber (a) and deposits on the anodic carbon brush (b) |
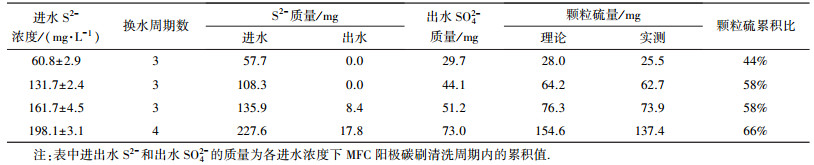
试验中未检测到SO32-, S2O32-也低于1 mg·L-1, 阳极S2-氧化的主要产物为S和SO42-.通过各清洗周期内的硫素平衡, 可计算实测颗粒硫量与S2-去除量的比值, 即颗粒硫累积比(表 3).发现颗粒硫累积比随着进水S2-总质量的增加而增加.这是因为增加进水S2-浓度可提高溶液电导率, 促进S2-通过电化学作用被氧化为颗粒硫, S2-提供的电子供体充足时还可抑制颗粒硫的进一步氧化.进水S2-浓度为(161.7±4.5) mg·L-1时的颗粒硫累积比可达58%, 继续增加进水S2-浓度可提高颗粒硫累积比, 但阳极出水S2-残余明显(每清洗周期残留17.8 mg).为避免阳极出水中的S2-进入阴极室后抑制阴极反应, 阳极进水S2-浓度不宜超过(161.7±4.5) mg·L-1.
| 表 3 不同进水S2-浓度下碳刷清洗周期内硫积累情况 Table 3 Sulfur accumulation during a washing interval of anodic carbon brush at different S2-concentrations |
1) 除硫硝化MFC可用于含S2-/NH4+废水的同步阳极除硫和阴极硝化, 通过非生物电化学作用和生物电化学作用共同产电, 非生物电化学产电量占MFC产电量的34.9%±7.1%.
2) 进水S2-浓度为(60.8±2.9)、(131.7±2.4)、(161.7±4.5) 和(198.1±3.1) mg·L-1时, 最佳阳极碳刷清洗周期分别为3、3、3、4个换水周期, 前3个浓度下的换水周期可分别缩短为6、8和8 h.
3) MFC阴极硝化完全, 不受进水S2-浓度影响, 但氧从阴极向阳极的渗漏导致阳极库仑效率较低( < 40%).适当增加进水S2-浓度有利于提高S2-去除负荷、颗粒硫累积比和产电性能.适宜的进水S2-浓度为(161.7±4.5) mg·L-1, 相应的S2-去除负荷、颗粒硫累积比、最大功率密度和周期产电量分别为(0.31±0.00) g·L-1·d-1、58%、5.77 W·m-3和(141.0±5.2) C.
4) MFC阳极碳纤维丝上聚积有明显的颗粒硫沉积物, 粒径约100 nm.阳极悬浮物与沉积物相比, 悬浮物中S0含量比例较高, 而S6+含量比例较低.
Cai J, Zheng P. 2013. Simultaneous anaerobic sulfide and nitrate removal in microbial fuel cell[J]. Bioresource Technology, 128: 760–764.
DOI:10.1016/j.biortech.2012.08.046
|
Cai J, Zheng P, Xing Y J, et al. 2015. Effect of electricity on microbial community of microbial fuel cell simultaneously treating sulfide and nitrate[J]. Journal of Power Sources, 281: 27–33.
DOI:10.1016/j.jpowsour.2015.01.165
|
Chae K J, Choi M, Ajayi F F, et al. 2008. Mass transport through a proton exchange membrane (nafion) in microbial fuel cells[J]. Energy & Fuel, 22(1): 169–176.
|
Freguia S, Tsujimura S, Kano K. 2010. Electron transfer pathways in microbial oxygen biocathodes[J]. Electrochimica Acta, 55(3): 813–818.
DOI:10.1016/j.electacta.2009.09.027
|
国家环境保护局. 2002. 水和废水监测分析方法(第4版)[M]. 北京: 中国环境科学出版社.
|
Guo Q, Hu H Y, Shi Z J, et al. 2016. Towards simultaneously removing nitrogen and sulfur by a novel process:Anammox and autotrophic desulfurization-denitrification (AADD)[J]. Chemical Engineering Journal, 297: 207–216.
DOI:10.1016/j.cej.2016.03.138
|
Jiang G, Sharma K R, Guisasola A, et al. 2009. Sulfur transformation in rising main sewers receiving nitrate dosage[J]. Water Research, 43(17): 4430–4440.
DOI:10.1016/j.watres.2009.07.001
|
Kim J R, Zuo Y, Regan J M, et al. 2008. Analysis of ammonia loss mechanisms in microbial fuel cells treating animal wastewater[J]. Biotechnology and Bioengineering, 99(5): 1120–1127.
DOI:10.1002/(ISSN)1097-0290
|
Lee C Y, Ho K L, Lee D J, et al. 2012. Electricity harvest from nitrate/sulfide-containing wastewaters using microbial fuel cell with autotrophic denitrifier, Pseudomonas sp.C27[J]. International Journal of Hydrogen Energy, 37(20): 15827–15832.
DOI:10.1016/j.ijhydene.2012.01.092
|
梁鹏, 范志明, 曹效鑫, 等. 2007. 微生物燃料电池表观内阻的构成和测量[J]. 环境科学, 2007, 28(8): 1894–1898.
|
Sun H, Xu S, Zhuang G, Zhuang X. 2016. Performance and recent improvement in microbial fuel cells for simultaneous carbon and nitrogen removal:A review[J]. Journal of Environmental Sciences, 39: 242–248.
DOI:10.1016/j.jes.2015.12.006
|
Sun M, Mu Z X, Chen Y P, et al. 2009. Microbe-assisted sulfide oxidation in the anode of a microbial fuel cell[J]. Environmental Science & Technology, 43(9): 3372–3377.
|
You S J, Ren N Q, Zhao Q L, et al. 2009. Improving phosphate buffer-free cathode performance of microbial fuel cell based on biological nitrification[J]. Biosensors Bioelectronics, 24(12): 3698–3701.
DOI:10.1016/j.bios.2009.05.015
|
Virdis B, Rabaey K, Yuan Z, et al. 2008. Microbial fuel cells for simultaneous carbon and nitrogen removal[J]. Water Research, 42(12): 3013–3024.
DOI:10.1016/j.watres.2008.03.017
|
Virdis B, Rabaey K, Rozendal R A, et al. 2010. Simultaneous nitrification, denitrification and carbon removal in microbial fuel cells[J]. Water Research, 44(9): 2970–2980.
DOI:10.1016/j.watres.2010.02.022
|
魏炎, 张少辉, 赵丽, 等. 2016. 反硝化脱硫微生物燃料电池的可行性研究[J]. 环境科学学报, 2016, 36(8): 2832–2837.
|
Xie S, Liang P, Chen Y, et al. 2011. Simultaneous carbon and nitrogen removal using an oxic/anoxic-biocathode microbial fuel cells coupled system[J]. Bioresource Technology, 102(1): 348–354.
DOI:10.1016/j.biortech.2010.07.046
|
于景荣, 衣宝廉, 韩明, 等. 2001. Nafion膜厚度对质子交换膜燃料电池性能的影响[J]. 电源技术, 2001, 25(6): 384–386.
|
远野, 王爱杰, 马素丽, 等. 2014. 反硝化脱硫工艺中生物硫分布特征及分离方法[J]. 哈尔滨工业大学学报, 2014, 46(8): 34–39.
DOI:10.11918/j.issn.0367-6234.2014.08.006 |
Zhang B, Zhao H, Shi C, et al. 2009. Simultaneous removal of sulfide and organics with vanadium (Ⅴ) reduction in microbial fuel cells[J]. Journal of Chemical Technology and Biotechnology, 84(12): 1780–1786.
DOI:10.1002/jctb.v84:12
|
Zhao F, Rahunen N, Varcoe J R, et al. 2008. Activated carbon cloth as anode for sulfate removal in a microbial fuel cell[J]. Environmental Science & Technology, 42(13): 4971–4976.
|