 2017, Vol. 37
2017, Vol. 37



2. 西南交通大学地球科学与环境工程学院, 成都 611756
2. Faculty of Geosciences and Environmental Engineering of Southwest Jiaotong University, Chengdu 611756
腐殖酸是一类广泛存在于土壤、江河、湖泊、沼泽和森林等自然环境中的高分子天然有机物(张爱平, 2008).腐殖酸不仅会影响土壤肥沃程度、植物生长状态及矿物质的积累和迁移, 而且也关系到环境生态平衡和人类健康(成绍鑫, 2007).但是, 在水环境中, 过量的腐殖酸却会影响水体的色度和浊度, 且可与水体中毒性因子等发生络合反应, 从而形成腐殖酸络合体, 能增加其生物毒性(Zhang et al., 2009);不仅如此, 腐殖酸还能在消毒过程中和含氯消毒剂反应生成具有“三致效应”的消毒副产物(Helal et al., 2011).特别是以垃圾渗滤液为典型代表的高浓度腐殖酸废水, 由于有机物浓度较高、可生化性较差和盐分含量较高便成为了水处理领域的研究热点, 目前, 垃圾渗滤液主要处理方法有:生化法、高级氧化(吴彦瑜等, 2010)、混凝(周勤等, 2002)和新型材料吸附(朱志平等, 2011)等技术.
高级氧化法是指以羟基自由基为主要氧化剂的氧化方法, 其中主要包括光催化氧化、臭氧氧化、芬顿氧化等.其中臭氧氧化法因具有高效、经济、操作简便等优点, 广大学者将其用于难降解有机废水的预处理以提升其可生化性.但张维佳等(2000)认为单独臭氧氧化法存在臭氧利用效率不高、污染物去除效果不佳等不足.而王利平等(2015)认为臭氧联合非均相催化技术能够促进臭氧向羟基自由基的转变, 从而提高臭氧利用率及有机物矿化效率, 加速废水中难降解有机物的氧化.一方面, 铁是一种过渡金属, 其核外具有较多的空轨道及较高的离子半径能强烈地吸附水分子, 表面羟基在水/氧化物表面与臭氧形成一系列的链式反应;另一方面, 铈作为常见的稀土元素, 常用作催化助剂改善其催化效果(何宏平等, 2015).故在工程应用方面, 负载活性碳-臭氧催化氧化技术在垃圾渗滤液、焦化废水等特种废水的处理中逐渐被认可, 但Fe-Ce/GAC催化臭氧降解高浓度降解腐殖酸类废水的研究鲜有, 特别是腐殖酸在单独臭氧和催化剂-臭氧体系的降解效能与机理值得深入探讨.
鉴于此, 本研究采用浸渍焙烧法将铁、铈元素负载到粒状活性炭, 用于催化臭氧降解高浓度腐殖酸, 以不同浸渍液及焙烧温度为主要影响因素, 分析不同条件下制备的催化剂对臭氧降解腐殖酸的催化效果和催化机理, 为垃圾渗滤液、垃圾浓缩液等含高浓度腐殖酸类废水的高效处理提供技术支撑.
2 材料与方法(Materials and methods) 2.1 试验材料试验水样:根据前期对四川省多个卫生填埋场的垃圾渗滤液和渗滤液浓缩液进行了调研, 其中, 渗滤液COD为2000~60000 mg·L-1, 腐殖酸浓度为300~2960 mg·L-1, 故模拟废水由商品腐殖酸(纯度≥90%)配置, 废水浓度为3.00 g·L-1, 并调节pH值至8.0左右, 过滤后待用.此时, 废水COD为1983.93 mg·L-1.
化学试剂:粒状活性炭;九水合硝酸铁(以下简称硝酸铁)、氢氧化钠、硝酸镧、硝酸铈等, 均为分析纯.
主要仪器:成都方舟科技pHS-25型酸度计、上海喆图TMF-4-13陶瓷马弗炉、上海谱元Alpha-1106分光光度计、日本JSM.5900LV扫描电子显微镜、北京普析XD-2型XRD仪.
实验装置:上海康特KT-OZ-5G型臭氧发生器1台, 自制有机玻璃圆柱形臭氧反应器1个, 有效高度为70 cm, 有效直径为4 cm, 实验装置如图 1所示.
 |
| 图 1 实验装置示意图(1.臭氧发生器; 2.圆筒形反应器; 3.填料柱; 4.催化剂; 5.曝气头; 6.空气管; 7.臭氧吸收瓶) Fig. 1 Schematics of experimental set-up |
活性炭改性:清洗浸泡活性炭至无杂质后, 再取5%氢氧化钠溶液和5%硝酸溶液分别浸泡活性炭12 h, 除去表面杂质, 再用去离子水洗涤至中性, 在105 ℃下干燥24 h后置于干燥器中备用, 记为GAC.
催化剂制备:常温下, 在50 mL蒸馏水中溶解设定浓度的无机铁盐、设定浓度的稀土元素、设定浓度的硝酸钾, 定容至100 mL待用.在浸渍液中加入20 g改性活性炭, 静态浸渍12 h后于80 ℃条件下烘干.烘干后的活性炭放入马弗炉于设定温度下保温焙烧3 h, 制得载铁催化剂.
催化臭氧实验:取出5.0 g催化剂, 将其浸泡于腐殖酸废水中12 h, 至吸附饱和, 预先消除吸附作用对污染物去除的影响.然后, 准确量取250 mL腐殖酸废水于自制反应器中, 取出预先消除吸附作用后的催化剂(占废水质量的2%)装于填料塔中, 使得催化剂与废水充分接触.在温度25 ℃、臭氧投量1.24 g·h-1, 反应时间40 min的条件下, 测定废水出水COD、腐殖酸浓度.
2.3 分析方法常用在波长254 nm处测得的吸光度, 以表征含腐殖酸废水中的腐殖酸含量(郑可, 2012; Yang et al., 2006), 且经测得的腐殖酸浓度的标准曲线如图 2所示, 其浓度与吸光度呈线性相关, 故腐殖酸浓度用UV254表征.采用微波密封消解-重铬酸钾法快速测定COD.
 |
| 图 2 腐殖酸浓度的标准曲线 Fig. 2 The calibration curve of humic acid |
用XRD仪检测, CuKα为射线源, 管电压为30 kV, 管电流为40 mA, 连续扫描, 扫描方式为θ/2θ.
电镜扫描及EDS扫描:电压为20 kV, 分辨率为3.0 nm.
3 试验结果与讨论(Results and discussion) 3.1 制备条件对腐殖酸降解效果的影响与分析 3.1.1 前驱体对腐殖酸降解效果的影响分别配置质量分数为2.0%的硝酸铁浸渍液、氯化铁浸渍液、硫酸亚铁浸渍液, 将其分别放入活性炭进行浸渍后焙烧, 并将制备的催化剂投入臭氧反应装置中进行催化实验, 不同条件下制备的催化剂对臭氧降解腐殖酸效果的影响如图 3所示.
 |
| 图 3 无机铁盐对催化臭氧效果的影响 Fig. 3 Influence of the inorganic ferric salt on the catalytic degradation efficiency of HA with ozone |
由图 3可知, 未投加催化剂的腐殖酸废水经臭氧氧化反应40 min后, 其COD、腐殖酸去除率仅17.1%、43.0%.在以上3种铁盐前驱体中, 经硝酸铁浸渍的催化剂催化效果最好, 其COD、腐殖酸去除率较未投加催化剂组高出11.2%和9.6%.由于硝酸铁溶液呈弱酸性, 低浓度硝酸具有打通活性炭内部及表面的孔洞结构的作用, 使活性炭孔容及比表面积增大, 有利于铁离子在活性炭中充分分散及均匀分布, 能有效减少活性物质的团聚(李伟峰等, 2006);负载的活性物质即铁氧化物能够促进臭氧转化为氧化能力更强的羟基自由基, 其表面羟基的形成能矿化有机物为二氧化碳和水(Jung et al., 2008; 张悦, 2015).另一方面, 焙烧过程中由硝酸铁分解释放的氮类氧化物可扩大活性炭的孔隙和比表面积, 使得催化效果进一步增加, 故后续试验选择硝酸铁作为前驱体.
3.1.2 焙烧温度对腐殖酸降解效果的影响>配置质量分数为2.0%的硝酸铁浸渍液, 分别设定焙烧温度为250、300、350、400、450 ℃.将制备的催化剂投加臭氧反应装置中进行催化实验, 焙烧温度对催化臭氧降解腐殖酸效果的影响如图 4所示.
 |
| 图 4 焙烧温度对催化臭氧效果的影响 Fig. 4 Influence of calcination temperature on the catalytic degradation efficiency of HA with ozone |
如图 4所示, 废水出水COD及腐殖酸去除率随催化剂焙烧温度先逐渐升高后减小, 其中350 ℃焙烧后的催化剂催化氧化废水效果最好, 其出水COD、腐殖酸去除率分别为28.3%、52.6%.在250~350 ℃的温度区间内, 随着焙烧温度的逐渐升高, 催化剂的催化性能越好.但随着催化剂焙烧温度的持续升高, 催化剂的催化效果反而逐渐降低.这是由于焙烧温度对于催化剂活性物质的形成十分关键, 在一定的温度范围内, 随着温度的逐渐升高, 催化剂进行活化和晶粒再分配, 能生成更多的碱性基团, 增强了活性炭表面极性, 更利于污染物和臭氧吸附进行催化反应(陆珍珍, 2011);另一方面, 随着温度的持续上升, 使得活性炭表面造成孔洞塌陷、孔径结构坍塌, 改变活性炭内部固有的孔洞结构, 降低了活性炭的吸附效果, 进而影响了催化剂的催化效果, 这也与李伟等(2004)的研究结果一致.
3.1.3 铁盐浓度对腐殖酸降解效果的影响配置不同质量分数的硝酸铁浸渍液(0.1%、0.5%、1.0%、2.0%、4.0%), 将活性炭进行浸渍后焙烧, 将制备的催化剂投加臭氧反应装置中进行催化实验, 不同条件下制备的催化剂对催化臭氧降解腐殖酸效果的影响如图 5所示.
 |
| 图 5 硝酸铁浓度对催化臭氧效果的影响 Fig. 5 Influence of ferric nitrate concentration on the catalytic degradation efficiency of HA with ozone |
由图 5可知, 随着浸渍液Fe(NO3)3浓度增加, 模拟废水中COD及腐殖酸去除率逐渐增加, 其中以浓度为2.0% Fe(NO3)3浸渍后所制成的催化剂进行臭氧催化氧化反应后, 其COD、腐殖酸去除率达28.3%和52.6%, 这是由于当浸渍液浓度升高时, 活性炭所吸附的铁离子不断增加;但是硝酸铁浸渍液浓度过高时, 在焙烧过程中, 高浓度的铁元素会以大颗粒结晶不稳定的状态存在于活性炭表面.活性炭具有很大的比表面积, 所以当物质的负载量低于其最大吸附值时, 其中物质的活性组分会以非结晶的无定型态或微晶形态存在于活性炭中, 从而表现出了极大的催化活性(张悦, 2015);而当负载量高于其表面极大吸附值时, 会形成大颗粒结晶态的氧化铁, 从而导致催化活性降低, 或者在洗涤和催化反应过程中以大颗粒结晶态掉落从而使得催化效果反而降低(Qi et al., 2016).
3.1.4 稀土元素种类对腐殖酸降解效果的影响分别配置含有质量分数为“1.0%的硝酸铈”、“1.0%硝酸铈+2.0%硝酸铁”、“1.0%硝酸镧”和“1.0%硝酸镧+2.0%硝酸铁”的浸渍液, 放入活性炭进行浸渍后焙烧, 将制备的催化剂投加臭氧反应装置中进行催化实验, 不同种类稀土元素对催化臭氧降解腐殖酸效果的影响如图 6所示.
 |
| 图 6 稀土元素种类对催化臭氧效果的影响 Fig. 6 Influence of rare earth type on the catalytic degradation efficiency of HA with ozone |
如图 6所示, 铁与稀土元素复合型催化剂均比单一稀土元素催化剂催化效果佳.其中, Fe-Ce催化剂组的COD、腐殖酸去除率高达37.4%、59.4%, 较其他催化剂的催化效果有较大幅度的提升.不仅是因为硝酸铁焙烧后产生的氧化铁具有催化作用, 而且铈的4f轨道可以有效的储存伴随氧空位所形成自由电子, 这些自由电子可以较好的促进分子氧的吸附氧化, 进而使得催化剂提高氧储存能力, 提高了其催化性能(詹望成等, 2012; Wu et al., 2016).邰佳等(2012)认为活性炭负载稀土元素后, 形成的活性组分有更小的粒径, 使得催化剂具有良好的量子及尺寸效应;另一方面, 添加硝酸铈的活性炭表面可以形成更加稳定的化学键.
3.1.5 Ce浓度对腐殖酸降解效果的影响配置含有质量2.0%硝酸铁、不同质量硝酸铈浸渍液(0.1%、1.0%、1.5%、2.0%、3.0%), 放入活性炭进行浸渍后焙烧, 将制备的催化剂投入臭氧反应装置中进行催化实验, 助剂硝酸铈浓度对催化臭氧降解腐殖酸效果的影响如图 7所示.
 |
| 图 7 硝酸铈浓度对催化剂催化效果的影响 Fig. 7 Influence of cerium nitrate concentration on the catalytic degradation efficiency of HA with ozone |
由图 7所示, 一定浓度条件下, 当Ce(NO3)3浓度升高时, 催化剂催化效果不断增加, 但硝酸铈浓度增加到1.5%以上时, 其催化效果反而降低.Ce(NO3)3浓度为1.5%时, 其COD、腐殖酸去除率分别为44.3%、63.9%.这是由于在浓度低于1.5%时, 活性炭并未吸附饱和, 且Carl等(2012)认为铈能够有效的促进羟基自由基的生成, 在一定条件下, 浸渍液含硝酸铈的量越多, 其制备的催化剂催化效果越好.而当浓度高于1.5%时, 活性炭近乎吸附饱和, 且助催化剂和催化剂已经达到较优配比, 催化效率达到较高水平, 而稀土元素浓度继续增高时, 可能会堵塞活性炭表面的部分孔道, 降低活性炭的比表面积, 使得催化剂吸附臭氧和水中有机污染物的能力降低;且在浓度较高的Ce(NO3)3浸渍液中, Ce(NO3)3和Fe(NO3)3会相互竞争活性炭的吸附位点, 反而导致活性炭中铁含量降低使得催化剂催化效果降低.
3.1.6 硝酸钾浓度对腐殖酸降解效果的影响配置含有质量2.0%硝酸铁、1.5%硝酸铈、设定浓度的硝酸钾浸渍液(0.1%、0.5%、1.0%、2.0%、4.0%), 投加活性炭进行浸渍后焙烧, 将制备的催化剂加入臭氧反应装置中, 不同条件下制备的催化剂对臭氧降解腐殖酸的效果如图 8所示.
 |
| 图 8 助剂硝酸钾浓度对催化臭氧效果的影响 Fig. 8 Influence of potassium nitrate concentration on the catalytic degradation efficiency of HA with ozone |
如图 8所示, 在一定的硝酸钾浓度范围内, 逐渐提高硝酸钾浓度会使得催化剂催化效果加强, 当硝酸钾浓度为1%时, 其COD、腐殖酸去除率分别为57.4%和74.8%, 较单独臭氧氧化腐殖酸废水, 其COD、腐殖酸去除率提高了40.3%、31.8%.当钾助剂浓度大于1%时, 其催化效果并没有明显的增强.这是由于添加钾助剂可以降低活性炭表面酸性, 减少副反应和活性成分的流失(张爱军等, 2008).与此同时, 钾助剂还可作为电子型助催化剂, 能提高催化剂催化性能;另一方面, 钾助剂可以有效减少金属氧化物的还原反应, 减少活性物质在催化过程中的消耗(张爱军等, 2008).
3.1.7 优化条件下制备的催化剂稳定性分析催化剂的使用寿命是评价催化剂的重要指标, 将3.1.6节所制备的催化剂进行催化氧化反应.反应后回收催化剂并烘干, 在相同的条件下重复用于催化臭氧降解腐殖酸, 催化剂的使用次数对催化臭氧降解腐殖酸效果的影响如图 9所示.
 |
| 图 9 催化剂使用次数对催化臭氧效果的影响 Fig. 9 Influence of the catalysts usage times on the catalytic degradation efficiency of HA with ozone |
如图 9所示, 当催化剂使用次数增加时, COD及腐殖酸去除率呈现略微下降趋势, 在使用5次之后, 由最初去除率57.4%、74.8%小幅下降至53.2%、65.7%.这是由于随着使用次数的增加, 有机物会进入活性炭的多孔介质中, 其孔容会被有机物堵塞, 导致活性炭孔容会逐渐变小, 覆盖了活性炭上的有效活性物质, 故臭氧与催化活性物质接触减少.在催化剂多次重复使用的情况下, 腐殖酸及COD的去除率受到的影响较小, 故所制得的催化剂依然具有较好的稳定性.
3.1.8 催化氧化腐殖酸类废水的工艺比较为对比催化剂的具体催化性能, 通过对其他臭氧催化剂的催化效果进行对比, 其中如表 1所示.
| 表 1 催化臭氧处理腐殖酸类废水比较 Table 1 Comparison of catalytic ozone treatment on humic substance wastewater |
由表 1可知, 催化剂能够极大的提高臭氧氧化效果, 如刘卫华等(2007)运用二价铜催化氧化降解渗滤液, 其TOC去除率提高了36.5%;而黄国忠等(2007)使用活性炭催化氧化降解腐殖酸, 腐殖酸的去除率仅仅上升了28.8%;王有乐等(2007)运用二氧化钛催化臭氧降解腐殖酸, 腐殖酸去除率由58%上升至86.8%;Gümüş等(2017) 使用铁包覆沸石和活性炭催化降解腐殖酸, 其DOC去除率提高了40.6%和26.7%.而就单独臭氧降解腐殖酸而言, 有机污染物的去除效果均不理想, 且仅在添加活性炭的催化条件下, 其催化降解腐殖酸的效果都较差, 有机物去除效果均有一定程度的提升.但是本催化剂通过在活性炭中负载Fe、Ce等活性物质, 使得废水的COD去除率提高了2.35倍, 腐殖酸去除率提高了0.74倍, 且还具有一定的重复利用性, 对比以上研究结果, 本文催化剂亦具有一定的研究价值和应用价值.
3.2 催化剂性能的表征与讨论 3.2.1 催化剂的SEM和EDS表征结果如图 10f所示, 通过EDS能谱分析了催化剂表面铁和铈的负载量, 其中, 铁、铈、钾的质量分数为8.85%、2.08%、1.38%, 活性炭中含有大量的活性催化成分.
 |
| 图 10 不同制备条件下催化剂的SEM和EDS谱(a.改性前活性炭; b.酸碱改性后活性炭; c.2.0%硝酸铁; d. 2.0%硝酸铁+1%硝酸铈; e. 2.0%硝酸铁+1.0%硝酸铈+1.0%硝酸钾; f. 2.0%硝酸铁+1.0%硝酸铈+1.0%硝酸钾) Fig. 10 SEM images and EDS spectra of the different catalysts |
其中图 10a、10b显示了活性炭改性前后表面形貌的变化, 未改性活性炭表面没有明显的孔洞结构, 改性后由于酸碱试剂打开了活性炭中的细小微孔使活性炭表面大孔体积增加, 比表面积明显增大, 使得活性物质负载量增加;图 10c显示, 经2.0%硝酸铁浸渍焙烧后的活性炭催化剂表面具有不规则金属光泽的细小颗粒状物质, 即负载的系列铁氧化物, 说明金属铁负载成功.而在此基础上, 浸渍液添加1.0%硝酸铈后(图 10d), 活性炭催化剂表面形成了含有金属光泽的颗粒状物且分布较均匀, 并未像图 10c那样排列紧密且无规则, 故稀土助剂能有效改善活性炭表面的团聚现象, 使得活性组分更加分散, 在后续催化臭氧氧化降解模拟废水反应中提高了接触几率, 使得其催化效果得到提升, 这与3.1.5节中结论一致.在此基础上, 浸渍液添加硝酸钾助剂后(图 10e), 可以看到活性炭表面具有金属光泽且活性组分紧密排列在活性炭表面, 较未添加硝酸钾时更加均匀且致密, 在催化臭氧氧化降解模拟废水的反应中使得臭氧与催化剂更好的接触, 从而提升了废水的处理效果, 这也与3.1.6节的结论高度吻合.
3.2.2 催化剂的XRD表征结果制备催化剂的XRD如图 11所示, 与XRD的JCPDS标准卡片对比可以看出, 原活性炭XRD图谱在衍射角为26.5°、38.0°处出现了较强的碳衍射峰.铁负载后的催化剂在衍射角为18.9°、28.9°和31.0°时有衍射峰出现, 故负载的活性物质为氧化铁.而负载铁和铈元素的活性炭中, 一方面, 衍射角为35.1°和28.7°有衍射峰出现, 其衍射峰为CeFeO3、Ce2O3的特征峰;另一方面, 负载铈后催化剂中活性物质的特征峰都有明显的减弱, 说明稀土元素可以改善活性物质在载体上的分散度, 在催化反应中促进了有机物的降解.
 |
| 图 11 不同制备条件下催化剂的XRD图谱 Fig. 11 XRD patterns of the catalysts |
稀土元素由于含有特殊的4f轨道使得其对臭氧具有良好的催化性质, 而CeO2是一种具有立方萤石型氧化物, 其孔隙较多, 可使反应体系中离子及臭氧快速分散(Trovarelli et al., 1997).由于Ce有三价和四价两种价态, 在反应过程中易发生氧化还原反应(邹兴等, 2000), 它们能在转化过程中使晶格中的氧脱离, 形成氧空位, 从而促使羟基自由基的生成;另一方面, 它们在转化的同时会形成含有氧缺陷结构CeO2-x(0<x<0.5) 的氧化物(式(1)), 这种非化学计量比的化合物可以形成化学吸附氧(式(2)~(3))从而提高催化剂的催化效果(Boaro et al., 2014).
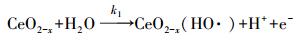
|
(1) |
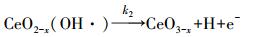
|
(2) |
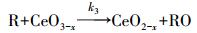
|
(3) |
此外, 在臭氧氧化反应过程中, 由于催化剂表面电荷并未平衡, 水中金属类氧化物如氧化铁会强烈吸附水分子, 其吸附的水分子会发生如下电离(式(4)), 电离生成的H+与OH-会与氧化铁产生表面羟基(Joseph et al., 2000).
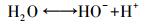
|
(4) |
溶解在水中的臭氧可与表面羟基发生反应生成羟基自由基.马军等(2005)认为羟基氧化铁可以可以产生大量的表面羟基进而催化臭氧产生大量的羟基自由基.其机理如式(5)~(7) 所示(Zhao et al., 2009):
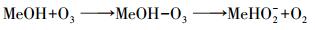
|
(5) |
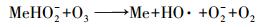
|
(6) |

|
(7) |
在催化臭氧降解腐殖酸的体系中, 由于助剂稀土元素Ce对臭氧的分散作用, 可以更快的对腐殖酸废水进行氧化, 提高臭氧利用率, 且Ce的氧化还原反应和水合氧化铁促进生成的羟基自由基是一种具有无选择性的氧化基团, 它较臭氧具有更高的氧化还原电位, 可以与更多的有机物如胡敏素和臭氧产生的中间产物发生取代、脱氢、电子转移或彻底的矿化反应, 从而提高了催化剂的催化效果.一方面, Ce产生的化学吸附氧具有的强氧化性, 能在催化剂表面通过吸附-降解的历程将腐殖酸废水中的有机物进行催化氧化反应;另一方面, CeO2在缺氧和富氧条件下会在Ce4+和Ce3+间不停转化, 形成氧空位(魏进, 2015), 该过程加速了腐殖酸降解速率, 故制备的Fe-Ce/GAC催化剂极大程度的提高了腐殖酸的降解速度.
4 结论(Conclusions)1) 利用在2.0%硝酸铁、1.5%硝酸铈、1.0%硝酸钾的混合溶液浸渍12 h, 经350 ℃焙烧3 h制备的Fe-Ce/GAC催化剂, 在pH值约8.0、温度为25 ℃、臭氧投量为1.24 g·h-1的条件下进行催化臭氧氧化, 反应40 min后模拟废水的COD、腐殖酸去除率较未投加催化剂的可提高40.3%、31.8%.
2) SEM图象显示活性炭经改性后利于活性物质的负载, 活性炭的孔容、比表面积较改性前有较大改变;EDS分析显示催化剂负载了大量的Fe、Ce等金属物质;XRD图谱显示制备的Fe-Ce/GAC催化剂含有Fe2O3、CeO2等多种活性物质, 添加了稀土元素的催化剂具有更好的分散度;优化条件下制备的催化剂具有较高的活性, 在重复使用5次后COD、腐殖酸去除率仅降低了4.2%、9.1%.
3) 催化剂的催化活性成分为Fe、Ce.在催化过程中CeO2进行氧化还原反应, 一方面提高了催化剂催化活性, 另一方面由CeO2-x氧化物形成化学吸附氧可对有机物进行吸附及氧化;而Fe2O3在臭氧氧化反应中首先生成羟基氧化铁, 羟基氧化铁与臭氧接触进而生成羟基自由基, 提高了臭氧的降解效率.后续可一方面展开实际废水中干扰因素(抗氧化剂或阴离子等)对催化剂稳定性能影响相的关工作;另一方面, 催化剂对废水中有机污染物的去除机制还可利用电子自旋共振技术对废水中自由基触发、链式反应和猝灭反应等基理论进行深入研究.
Boaro M, Giordano F, Recchia S, et al. 2014. On the mechanism of fast oxygen storage and release in ceria-zirconia model catalysts[J]. Applied Catalysis B, 52(3): 225–237.
|
Carla A O, Jose J M, Manuel F R, et al. 2012. Ceria and cerium-based mixed oxides as ozonation catalyst[J]. Chemical Engineering Journal, 200-202: 499.
DOI:10.1016/j.cej.2012.06.088
|
成绍鑫. 2007. 腐殖酸类物质概论[M]. 北京: 化学工业出版社.
|
Gümüş D, Akbal F. 2017. A comparative study of ozonation, iron coated zeolite catalyzed ozonation and granular activated carbon catalyzed ozonation of humic acid[J]. Chemosphere, 174: 218–231.
DOI:10.1016/j.chemosphere.2017.01.106
|
Jung H, Kim J W, Choi H, et al. 2008. Synthesis of nanosized biogenic magnetite and comparison of its catalytic activity in ozonation[J]. Applied Catalysis B:Environmental, 83(3): 208–213.
|
何宏平, 吴德礼, 马鲁铭, 等. 2015. 改性黄铁矿烧渣催化臭氧氧化水中活性黑5[J]. 同济大学学报(自然科学版), 2015, 43(11): 1728–1734.
DOI:10.11908/j.issn.0253-374x.2015.11.018 |
Helal A A, Murad G A, Helal A A. 2011. Characterization of different humic materials by various analytical techniques[J]. Arabian Journal of Chemistry, 4(1): 51–54.
DOI:10.1016/j.arabjc.2010.06.018
|
黄国忠, 丁月红, 董晓伟, 等. 2007. 活性炭催化臭氧氧化去除水中的腐殖酸[J]. 化工环保, 2007, 27(3): 200–203.
|
Joseph Y, Ranke W, Weiss W. 2000. Water on FeO(Ⅲ) and Fe3O4(Ⅲ):adsorption behavior on different surface terminations[J]. Journal of Physical Chemistry B, 104(14): 3224–3236.
DOI:10.1021/jp9932012
|
刘卫华, 季明, 张昕, 等. 2007. 催化臭氧氧化去除垃圾渗滤液中难降解有机物的研究[J]. 环境化学, 2007, 26(1): 58–61.
|
李伟峰, 祝社民, 宋天顺, 等. 2006. 负载铜活性炭催化剂制备及催化氧化印染废水[J]. 林产化学与工业, 2006, 26(4): 25–30.
|
李伟, 孙德智, 刘长安, 等. 2004. 活性炭负载复合催化剂分解臭氧的研究[J]. 哈尔滨工业大学学报, 2004, 36(5): 624–630.
|
陆珍珍. 2011. 负载铈活性炭催化臭氧化氯霉素研究[D]. 广州: 广州大学
http://cdmd.cnki.com.cn/article/cdmd-11078-1012262612.htm |
马军, 张涛, 陈忠林, 等. 2005. 水中羟基氧化铁催化臭氧分解和氧化痕量硝基苯的机理探讨[J]. 环境科学, 2005, 26(2): 78–82.
|
Qi F, Chu W, Xu B. 2016. Comparison of phenacetin degradation in aqueous solutions by catalytic ozonation with CuFe2O4 and its precursor:surface properties, intermediates and reaction mechanisms[J]. Chemical Engineering Journal, 284: 28–36.
DOI:10.1016/j.cej.2015.07.095
|
邰佳, 刘勇健. 2012. 稀土催化臭氧氧化法降解印染废水的研究[J]. 环境科学与技术, 2012, 35(10): 171–181.
DOI:10.3969/j.issn.1003-6504.2012.10.037 |
Trovarelli A, De L C, Dolcetti G. 1997. Design better cerium-based oxidation catalysts[J]. Chemical Technology, 27(6): 32–37.
|
王有乐, 杨艳丽, 王玉双, 等. 2007. 负载型TiO2催化臭氧化去除腐殖酸的实验研究[J]. 环境科学与技术, 2007, 30(11): 35–37.
DOI:10.3969/j.issn.1003-6504.2007.11.013 |
王利平, 沈肖龙, 倪可, 等. 2015. 非均相催化臭氧氧化深度处理炼油废水[J]. 环境工程学报, 2015, 9(5): 2297–2302.
|
魏进. 2015. 氧化铝负载纳米铜基脱硫剂的制备及其脱硫效能的分析[D]. 哈尔滨: 哈尔滨理工大学
http://cdmd.cnki.com.cn/Article/CDMD-10214-1015576154.htm |
Wu D, Liu Y, He H, et al. 2016. Magnetic pyrite cinder as an efficient heterogeneous ozonation catalyst and synergetic effect of deposited Ce[J]. Chemosphere, 155: 127–134.
DOI:10.1016/j.chemosphere.2016.04.041
|
吴彦瑜, 郑可, 陈东宇, 等. 2010. Fenton试剂氧化降解腐殖酸动力学[J]. 环境科学, 2010, 31(9): 2085–2091.
|
Yang J K, Lee S M. 2006. Removal of Cr(Ⅵ) and humic acid by using TiO2 photocatalysis[J]. Chemosphere, 63(10): 1677–1684.
DOI:10.1016/j.chemosphere.2005.10.005
|
张爱平. 2008. 基于准好氧矿化垃圾床的垃圾渗滤液处理研究[D]. 成都: 西南交通大学
|
张爱军, 武红丽, 曹飞, 等. 2008. 钾助剂对硼铝酸铜催化脱氢环化性能的影响[J]. 分子催化, 2008, 22(5): 434–438.
|
Zhang L, Li A, Lu Y, et al. 2009. Characterization and removal of dissolved organic matter (DOM) from landfill leachate rejected by nanofiltration[J]. Waste Management, 29(3): 1035–1040.
DOI:10.1016/j.wasman.2008.08.020
|
张维佳, 王宝贞, 伍悦滨. 2000. 臭氧及深度氧化法去除水中污染物[J]. 给水排水, 2000, 26(5): 15–18.
|
张悦. 2015. O3/MgO催化氧化对苯酚的降解行为研究[D]. 成都: 西南石油大学
http://cdmd.cnki.com.cn/Article/CDMD-10615-1015599148.htm |
Zhao L, Ma J, Sun Z, et al. 2009. Mechanism of heterogeneous catalytic ozonation of nitrobenzene in aqueous solution with modified ceramic honeycomb[J]. Applied Catalysis B:Environmental, 89(3): 326–334.
|
詹望成, 郭耘, 郭杨龙, 等. 2012. 稀土催化材料的制备、结构及催化性能[J]. 中国科学:化学, 2012, 42(9): 1289–1307.
|
郑可, 周少奇, 杨梅梅. 2012. 臭氧降解高浓度腐殖酸动力学[J]. 环境科学, 2012, 33(3): 879–884.
|
周勤, 肖锦, 吴友明. 2002. PASS混凝去除给水腐殖酸的研究[J]. 环境科学与技术, 2002, 25(6): 8–10, 47.
|
朱志平, 黄可龙, 周艺. 2011. 碳纳米管吸附腐殖酸的动力学、热力学及机理研究[J]. 无机材料学报, 2011, 26(2): 170–174.
|
邹兴, 卢惠民, 方克明. 2000. 变价稀土氧化物对催化剂性能的影响[J]. 稀土, 2000, 21(2): 16–18.
|