 2017, Vol. 37
2017, Vol. 37



2. 中国科学院生态环境研究中心, 环境水质学实验室, 北京 100085
2. Key Laboratory of Aquatic Science and Technology, Research Center for Eco-Environmental Sciences, Chinese Academy of Sciences, Beijing 100085
随着水质排放标准的日益提高, 人们在优化传统水处理工艺的同时, 也在寻求与开发新的水处理技术.高级氧化技术(Advanced Oxidation Processes, AOPs)受到了日益重视.高级氧化技术是20世纪70年代以来在H2O2、O3、UV等单一氧化的技术基础上发展起来的新型化学氧化技术(Huang et al., 1993).其主要包括光催化氧化(PC)、电化学氧化(EC)、Fenton氧化、UV/H2O2氧化和臭氧氧化等(张旋等, 2009).而对于高浓度污染物来说, 单一氧化技术的降解效率有待提高.
根据不同高级氧化技术的特点, 将技术加以优化整合来处理污染物成为发展趋势.将电化学氧化与光催化氧化相结合的光电催化氧化技术(PEC)可以有效促进光生电子和空穴的分离, 进而高效去除有机污染物(Zhao et al., 2014);Brillas等(1999) 以铁为阳极氧化产生亚铁离子, 而以碳-聚四氟乙烯空气电极为阴极产生H2O2, 形成类Fenton反应来处理水体中的污染物质等.
本实验利用双电源体系, 以低压汞灯提供UV光, 利用RuO2/Ti为电阳极产生O2, 同时以ACF为阴极材料原位还原O2为体系提供大量H2O2, 在光源另一侧以高性能的TiO2/Ti为光阳极, 使光能利用率最大化.将EC、PEC及UV/H2O2氧化3种处理方式融为一体, 以高浓度的EDTA溶液为目标污染物, 研究阴极原位产生H2O2强化EC与PEC的效率与机制.本论文详细考察了电流密度、pH、曝气等因素对于体系降解EDTA效果的影响, 研究了降解反应机制, 并对能耗进行了分析.
2 实验部分(Experimental section) 2.1 实验装置实验装置如图 1所示.电源Ⅰ中:阳极为RuO2/Ti, 阴极为缚在RuO2/Ti表面的ACF;电源Ⅱ中:阳极为TiO2/Ti, 阴极材料为不锈钢, 电极面积均为25 cm2(5 cm×5 cm), 极板厚度1 mm.反应器中心插入低压汞灯提供UV光, 紫外灯功率为9 W, 紫外光波长λ= 254 nm.反应容器尺寸为6 cm×7 cm×9 cm, 实际反应液体积250 mL.电极板及紫外灯均利用长度为10 cm的绝缘夹板进行固定.实验采用直流电源(北京大华电子有限公司, AMRFL model:LPS302A 35 V/10 A)为体系提供电流.
 |
| 图 1 反应装置示意图 (1.碳纤维;2.RuO2/Ti;3.TiO2/Ti;4.不锈钢;5.低压汞灯;6.石英套管;7.磁子;8.EDTA溶液;9.220V交流电;10.搅拌器;11.暗箱;12.直流电源) Fig. 1 Schematic diagram of experimental set up (1.ACF, 2.RuO2/Ti, 3.TiO2/Ti, 4.stainless steel, 5.UV-light, 6.quartz tube, 7.magneton, 8.solution of EDTA, 9.alternating current of 220V, 10.stirrer, 11.camera box, 12. DC electrical source) |
Ti及RuO2/Ti电极片(北京恒力钛公司售)、TiO2/Ti的制备参考(Shang et al., 2003)、EDTA-2Na(C10H14N2Na2O8·2H2O, AR)、碘化钾(KI, AR)、无水乙酸钠(CH3COONa, AR)、硫氰酸铵(NH4SCN, AR)、十二水合硫酸铁铵(NH4Fe(SO4)2·12H2O, AR)、氢氧化钠(NaOH, AR)、邻苯二甲酸氢钾(C8H5KO4, GR)、盐酸(HCl, GR)、硫酸(H2SO4, GR)等, 羟基自由基(·OH)捕获剂为5, 5-dimethyl-1-pyrroline-N-oxide(DMPO)(C6H11NO, Sigma Chemical Corporation).其中硫酸铁铵溶液由0.5 mol·L-1的HCl溶液配制而成, 其余均用超纯水配制.超纯水由UPW-30型超纯水机提供.
2.3 分析方法及仪器EDTA浓度的测定:EDTA的测定采用显色法.取1 mL待测EDTA溶液于10 mL比色皿中, 依次加入0.8 mL乙酸钠缓冲溶液(0.4 g·L-1), 1 mL硫酸铁铵溶液(0.05 g·L-1), 1.5 mL硫氰酸铵溶液(3.45 mol·L-1), 用超纯水定容至10 mL, 充分摇匀后静置10 min, 利用紫外可见分光光度仪(U-3010, 日立公司)于λ=485 nm处测定吸光度.
H2O2浓度的测定:取1.50 mL待测溶液于石英比色皿中, 依次加入0.75 mL邻苯二甲酸氢钾(0.1 mol·L-1)和0.75 mL KI溶液(0.4 mol·L-1)溶液, 混合均匀, 静置2 min, 于λ=352 nm处测定吸光度, 该方法的检测限为10-6 mol·L-1(Ge et al., 2004).
本实验采用PHS-3E型pH计(雷磁)测定溶液pH;利用雷磁DZB-718型便携式多参数分析仪测定溶液中溶解氧;通过TOC-VCPH型(日本岛津公司)TOC分析仪测定溶液TOC变化;·OH采用电子顺磁共振波谱法测量(ESP 300E, 瑞士Bruker)(Gao et al., 2013).
3 结果与讨论(Results and discussion) 3.1 不同途径处理效果对比在相同的反应条件下, 研究了不同降解途径对EDTA降解效率的影响.实验结果如图 2a所示, 反应90 min 后, UV氧化、PC、PEC、EC、EC+UV/H2O2及PEC+EC+UV/H2O2这6种降解方式对EDTA的去除率分别为22.50%、24.34%、39.90%、62.32%、80.10%和90.80%, 可以看出多过程协同作用对EDTA的降解效率有明显提高.对反应前后溶液TOC进行测定, 结果如图 2b所示, PEC+EC+UV/H2O2体系对TOC的去除率最高, 可达43.70%.
 |
| 图 2 不同降解途径对EDTA的降解(a)及TOC的去除(b) ([EDTA]= 300 mg·L-1;[Na2SO4]= 0.075 mol·L-1;电流密度= 12 mA·cm-2;光电流密度= 0.012 mA·cm-2;pH=4.84) Fig. 2 The degradation rate of EDTA (a) and the degradation rate of TOC (b) though different degradation pathways ([EDTA]=300 mg·L-1; [Na2SO4]=0.075 mol·L-1; current density= 12 mA·cm-2; current density of PEC=0.012 mA·cm-2; pH= 4.84) |
这是由于阳极RuO2/Ti在氧化污染物的同时为体系提供大量O2(1) , 溶液中的O2在ACF阴极还原作用下生成大量H2O2(2) (Gong et al., 2016), H2O2在紫外灯的作用下产生氧化性极强的·OH(3) .在光源另一侧的电源Ⅱ中, 以性能良好的TiO2/Ti作为光阳极, 当紫外光照射TiO2/Ti阳极板时, 价带电子被激发跃迁至导带产生空穴, 夺取水分子的电子生成·OH, 光生电子在外加偏压的作用下, 有效地减少了光生电子与空穴的复合.因此本体系可在不外加O2及催化剂的前提下, 快速、高效的降解溶液中的EDTA.
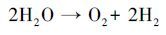
|
(1) |
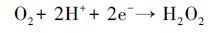
|
(2) |
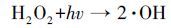
|
(3) |
由于ACF具有巨大的比表面积, 对EDTA会有一定的吸附作用, 为排除ACF的吸附作用对于体系降解EDTA的干扰, 因此在降解实验前对ACF进行了多次吸附实验.如图 3所示, c(EDTA)0= 300 mg·L-1, 单次吸附时长360 min.第1次循环中, 在ACF的吸附作用下, EDTA的去除率约为20%, 随着吸附循环次数的增加, EDTA的去除率明显减弱且趋于稳定, 此时ACF已达到吸附饱和, 表明活性炭纤维所产生的吸附作用已消除.
 |
| 图 3 不同循环次数下ACF阴极对EDTA的吸附 ([EDTA]= 300 mg·L-1;s(ACF)= 50 cm2) Fig. 3 The cathode ACF adsorbed EDTA in different batch cycles ([EDTA]=300 mg·L-1; s(ACF)= 50 cm2) |
电流密度是影响EC过程及原位还原O2生成H2O2的重要条件, 因此在光电流密度、pH、污染物浓度等条件相同的前提下, 考察了电流密度对于污染物去除的影响, 如图 4a所示.由图可以看出, 随着电流密度的增大, EDTA降解速率有一定提高.当电流密度从6 mA·cm-2提高到24 mA·cm-2, 反应90 min后, EDTA的降解率从85%提升到100%.由于随着电流密度的增加, 电阳极在加快污染物氧化速率的同时增强了析O2作用, 体系生成H2O2的浓度增加, 在UV光照射下产生的·OH增加, 因此降解速率加快.
 |
| 图 4 电流密度对EDTA去除率的影响(a)及不同初始pH对于EDTA去除率的影响(b) (a:[EDTA]= 300 mg·L-1;[Na2SO4]= 0.075 mol·L-1;光电流密度= 0.012 mA·cm-2;pH= 4.84; b:[EDTA]= 300 mg·L-1;[Na2SO4]= 0.075 mol·L-1;电流密度= 12 mA·cm-2;光电流密度= 0.012 mA·cm-2) Fig. 4 Effect of current density and initial pH on EDTA removal rate (a: [EDTA]= 300 mg·L-1; [Na2SO4]= 0.075 mol·L-1; current density of PEC= 0.012 mA·cm-2; pH= 4.84; b: [EDTA]= 300 mg·L-1; [Na2SO4]= 0.075 mol·L-1; current density= 12 mA·cm-2; current density of PEC=0.012 mA·cm-2) |
溶液pH决定了体系中H+的含量, 进而影响EDTA的降解速率.图 4b可看出, 当溶液pH值为碱性时, EDTA的降解速率相比减小.这是由于碱性条件会抑制反应式(2) 的进行, 且H2O2会以HO2·的形式存在(Sheng et al., 2011).但本体系为多途径联合作用, 为此EDTA的降解效率减小并不明显;当溶液初始pH为2.01时, EDTA的去除率达到90%所需时间为60 min, EDTA的降解速率显著提高.根据反应式(2) , 随着H+浓度的增加, H2O2的生成量增大.因此溶液在酸性条件下降解效果更为优异.
3.3 反应过程溶解氧的变化及曝气对降解实验的影响根据H2O2的生成条件可知, 溶液中O2的浓度对其影响较大.特测定了在不同反应条件下, 体系溶解氧的变化情况如图 5a所示.由图看出, 在RuO2/Ti阳极的析O2作用下, EC过程反应20 min后, 体系的溶解氧由3.20 mg·L-1迅速升至6.37 mg·L-1;而在PEC+EC+UV/H2O2体系下反应20 min后, 体系溶解氧仅上升到5.10 mg·L-1, 且随着反应时间的延长溶液溶解氧呈下降趋势.由此看出在RuO2/Ti阳极的析O2作用下, 溶液中溶解氧浓度逐渐提高, 并且在ACF阴极的还原作用下, 消耗溶液部分溶解转化为H2O2.
 |
| 图 5 反应过程溶解氧的变化(a)及曝气条件对EDTA去除率的影响(b) ([EDTA]= 300 mg·L-1;[Na2SO4]= 0.075 mol·L-1;电流密度= 12 mA·cm-2;光电流密度= 0.012 mA·cm-2;pH=4.84;O2:100 mL·min-1;N2:100 mL·min-1) Fig. 5 The changes of dissolved oxygen (a) and effect of aeration on EDTA removal rate (b) ([EDTA]=300 mg·L-1; [Na2SO4]=0.075 mol·L-1; current density= 12 mA·cm-2; current density of PEC= 0.012 mA·cm-2; pH= 4.84; O2: 100 mL·min-1; N2: 100 mL·min-1) |
同时考察了不同曝气条件对于EDTA降解效果的影响.如图 5b所示, 在其他条件相同时, 向体系通入O2或N2, EDTA的降解率无明显变化.由于不同降解方式的联合作用, 在阳极强烈的析O2作用下, 通入N2时并不能将体系内全部溶解氧排除;当向体系中通入O2时, 高浓度的溶解氧会促进ACF阴极还原O2生成H2O2.但当H2O2较高时, 部分H2O2会被阳极氧化分解, 如反应式(4) 、(5) 、(6) (Wang et al., 2011), 且H2O2与·OH也会发生副反应, 如反应式(8) (Borràs et al., 2010).为此本实验在不外加O2的条件下, 即可快速降解EDTA.

|
(4) |
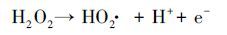
|
(5) |
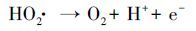
|
(6) |
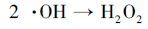
|
(7) |

|
(8) |
对于UV/H2O2强化PEC、EC的处理效果, H2O2的浓度尤为重要.在此考察了不同电流密度下的H2O2的产量及·OH的信号强度, 如图 6所示.由图 6a看出, H2O2的浓度随着电流密度增加而增大, 该结果与图 4a相吻合.随着反应的进行, H2O2的浓度趋于稳定状态.这是由于当溶液中H2O2达到一定浓度后, 会在阳极表面(异相过程)和溶液中(均相过程)发生化学分解(9) (Wang et al., 2008; Brillas et al., 2015), 也会被阳极氧化生成HO2·(10) , 并继续分解成O2(11) (Brillas et al., 2000), 所以稳态浓度时电化学生成的H2O2与发生分解的H2O2大致相等, 其浓度基本保持恒定.
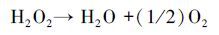
|
(9) |
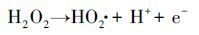
|
(10) |
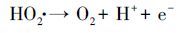
|
(11) |
图 6b看出, 当电流密度从6 mA·cm-2增大到12 mA·cm-2时, ·OH的峰值增加一倍.在紫外光的照射下, 随着H2O2浓度的增加, 其转化为·OH的浓度逐渐变大(3) .当电流密度达到一定值后, H2O2与·OH会发生副反应, 因此在电流密度为18 mA·cm-2、24 mA·cm-2时, ·OH的峰值无明显变化.
 |
| 图 6 不同电流密度对H2O2的产生量(a)及·OH浓度的影响(b) ([EDTA]= 300 mg·L-1;[Na2SO4]= 0.075 mol·L-1;光电流密度= 0.012 mA·cm-2;pH=4.84) Fig. 6 Effect of current density on H2O2 (a) and ·OH production (b) ([EDTA]=300 mg·L-1; [Na2SO4]=0.075 mol·L-1; PowerⅡ: current density of PEC= 0.012 mA·cm-2; pH= 4.84) |
在UV氧化、PC、PEC、EC、EC+UV/H2O2及PEC+EC+UV/H2O2降解EDTA的过程中, 能耗主要包括两组电极板和紫外灯的电能消耗.根据不同降解方式, 通过计算降解1 g污染物EDTA所消耗的电量, 来分析体系降解过程中的能耗需求.计算公式见式(12) .

|
(12) |
式中:W为能耗(kWh·g-1);U为电源电压(V);I为电源电流(A);P为紫外灯功率(W);h为反应时间(h);Δx为EDTA降低值(g·L-1).
根据公式(12) , 计算在电流密度和光电流密度分别为12 mA·cm-2和0.012 mA·cm-2, 电压分别为3.5 V和0.008 V的条件下, UV氧化、PC、PEC、EC、EC+UV/H2O2及PEC+EC+UV/H2O2这6种方式的能耗情况, 如表 1所示.
| 表 1 不同过程降解EDTA能耗分析 Table 1 Unit energy consumption and EDTA removal under different ways |
1) 通过对比UV氧化、PC、PEC、EC、EC+UV/H2O2及PEC+EC+UV/H2O2降解EDTA的去除效率得出:在反应条件相同的情况下, 反应90 min后EDTA的去除率分别为22.50%、24.34%、39.90%、62.32%、80.10%及90.80%.本实验利用PEC、EC和UV/H2O2氧化3种氧化形式的共同作用, 有效实现了水中难降解有机污染物的高效去除.
2) 通过控制溶液初始pH、电流密度、溶解氧等反应条件, 探讨了影响EDTA降解效率的因素.在溶液为酸性, 电流密度为0.012 A·cm-2, 光电流密度为 0.012 mA·cm-2及较高溶解氧的条件下, EDTA的降解效率较快, 且随着电流密度的增加, H2O2的产量及·OH浓度均有所增加, EDTA的降解效率增大.
3) 本体系利用PEC、EC及UV/H2O2的多过程共同作用, 可在不外加O2及任何药剂的前提下, 产生高浓度H2O2及·OH, 对于水中EDTA的降解效果明显, 能耗较低, 应用前景广阔.
| [${referVo.labelOrder}] | Borràs N, Oliver R, Arias C, et al. 2010. Degradation of atrazine byelectrochemical advanced oxidation processes using a boron-doped diamondanode[J]. American Chemical Society, 114(24) : 6613–6621. |
| [${referVo.labelOrder}] | Brillas E, C A, Martínez-Huitle C A. 2015. Decontamination of wastewaters containing synthetic organic dyes by electrochemical methods[J]. An updated review[J]. Applied Catalysis B:Environmental, 166-167 : 603–643. DOI:10.1016/j.apcatb.2014.11.016 |
| [${referVo.labelOrder}] | Brillas E, Sauleda R, Casado J. 1999. Use of an acidic Fe/O2 cell for wastewater treatment:degradation of aniline[J]. Journal of the Electrochemical Society, 146(12) : 4539–4543. DOI:10.1149/1.1392671 |
| [${referVo.labelOrder}] | Enric B, Calpe J C, Casado J. 1999. Mineralization of 2,4-D by advanced electrochemical oxidation processes[J]. Water Research, 34(8) : 2253–2262. |
| [${referVo.labelOrder}] | Gao Y, Zhou Y, Wang H, et al. 2013. Simultaneous Silver Recovery and Cyanide Removal from Electroplating Wastewater by Pulse Current Electrolysis Using Static Cylinder Electrodes[J]. Industrial & Engineering Chemistry Research, 52(17) : 5871–5879. |
| [${referVo.labelOrder}] | Ge J, Qu J. 2004. Ultrasonic irradiation enhanced degradation of azo dye on MnO2[J]. Applied Catalysis B:Environmental, 47(2) : 133–140. DOI:10.1016/j.apcatb.2003.08.001 |
| [${referVo.labelOrder}] | Gong Y, Li J, Zhang Y, et al. 2016. Partial degradation of levofloxacin for biodegradability improvement by electro-Fenton process using an activated carbon fiber felt cathode[J]. J Hazard Mater, 304 : 320–328. DOI:10.1016/j.jhazmat.2015.10.064 |
| [${referVo.labelOrder}] | Huang C P, Dong C, Tang Z. 1993. Advanced chemical oxidation:its present role and potential future in hazardous waste treatment[J]. Waste Management, 13(5) : 361–377. |
| [${referVo.labelOrder}] | Shang J, Li W, Zhu Y F. 2003. Structure and photocatalytic characteristics of TiO2 film photocatalyst coated on stainless steel webnet[J]. Journal of Molecular Catalysis A-Chemical, 202(1) : 187–195. |
| [${referVo.labelOrder}] | Sheng Y P, Song S L, Wang X L, et al. 2011. Electrogeneration of hydrogen peroxide on a novel highly effective acetylene black-PTFE cathode with PTFE film[J]. Electrochimica Acta, 56(24) : 8651–8656. DOI:10.1016/j.electacta.2011.07.069 |
| [${referVo.labelOrder}] | Wang A, Li Y Y, Estrada A L. 2011. Mineralization of antibiotic sulfamethoxazole by photoelectro-Fenton treatment using activated carbon fiber cathode and under UVA irradiation[J]. Applied Catalysis B:Environmental, 102(3/4) : 378–386. |
| [${referVo.labelOrder}] | Wang A, Qu J, Liu H, et al. 2008. Mineralization of an azo dye Acid Red 14 by photoelectro-Fenton process using an activated carbon fiber cathode[J]. Applied Catalysis B:Environmental, 84(3/4) : 393–399. |
| [${referVo.labelOrder}] | Zhao X, Guo L, Qu J. 2014. Photoelectrocatalytic oxidation of Cu-EDTA complex and electrodeposition recovery of Cu in a continuous tubular photoelectrochemical reactor[J]. Chemical Engineering Journal, 239 : 53–59. DOI:10.1016/j.cej.2013.10.088 |
| [${referVo.labelOrder}] | 张旋, 王启山. 2009. 高级氧化技术在废水处理中的应用[J]. 水处理技术, 2009, 35(3) : 18–22. |