 2016, Vol. 36
2016, Vol. 36



元素碳示踪物法(The EC tracer method)主要用于有机碳的一次和二次来源估算.该方法最早是针对元素碳(elemental carbon,EC)/有机碳(organic carbon,OC)元素分析仪所测量得到的颗粒相中的EC和OC开发的(Turpin and Huntzicker,1991).因为其仅利用OC和EC的测量数据,方法较为简单,被广泛的应用于二次有机碳的估算当中(Turpin and Huntzicker,1995;Lim and Turpin,2002a;Lim and Turpin,2002b;Lim et al.,2003).元素碳示踪物法的原理介绍如下:燃烧源是EC的主要排放来源,而OC来源较为复杂,包括一次燃烧源排放的OC(OCpri)、二次生成(OCsec)和非燃烧源直接排放的OC以及背景值(OCnon).
EC主要来自一次燃烧源排放,没有二次转化的来源.因此如果大气一次源排放OC/EC的比值以及非燃烧源直接排放的OC满足以下两个假设(Turpin and Huntzicker,1995):
(1) 碳质气溶胶的一次排放源比较固定,源强变化也不大,在时间和空间上有较好的重现性;
(2) OC和EC主要来自于燃烧源,非燃烧源直接排放的OC所占比重很小,可忽略或为一个常数.
则可以利用EC浓度乘以一次燃烧源中OC/EC的比值,得到测量站点处大气中的一次有机碳浓度OCpri.总有机碳OCtotal和OCpri的差值即二次有机碳浓度(OCsec).公式如下:
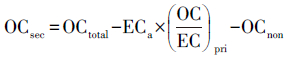
|
(1) |
元素碳示踪物法也被广泛应用于我国大气有机碳中二次来源的估算.Lin等(2009)估算了北京四季二次有机碳(secondary organic carbon,SOC)比例约在19%~45%之间,其中冬天比例最低,夏季最高.Cao等(2007)报道了全国14个城市的OC/EC浓度值,发现观测期间二次有机碳平均占OC总浓度的31.7%.利用元素碳示踪物法估算SOC的贡献,我国经济发展迅速的珠江三角洲城市地区和京津唐地区SOC约占OC的33%~62%(Cao et al.,2004;Duan et al.,2007;Xiao et al.,2011;Feng et al.,2012).
虽然元素碳示踪物法目前被广泛的应用于一次和二次有机碳的源解析工作,但该方法仍具有较大的不确定性,因为大部分情况下OC/EC的排放速率随着源的变化而变化,因此OC/EC在一次源中的比值 [(OC/EC)pri]随时间不是一个常数,并且排放源不同,OC/EC的比值亦不同,特别是在一次排放源复杂的城市地区.同时大气中的有机气溶胶受到二次源生成的影响,OC和EC在一次源中的比值容易被高估,从而一次有机碳(Primary organic carbon,POC)浓度容易被高估,而SOC浓度被低估.例如,Zhang等(2005)同时利用受体模型和元素碳示踪物法解析美国匹兹堡有机气溶胶来源,发现元素碳示踪物法估算得到的SOC浓度较受体模型估算结果偏低约30%.类似的结论也在其它地区的源解析方法比对中发现(Xiao et al.,2011;Hu et al.,2012).因此,得到准确的有机碳和元素碳在一次源中的比值是准确区分一次源和二次源对有机气溶胶贡献的关键.
目前比较常用的计算(OC/EC)pri方法包括:①利用排放清单得到一次排放的OC/EC比值(Docherty et al.,2008).②利用以一次源排放主要的时间段内的OC/EC的比值(Turpin and Huntzicker,1991; 1995);③光化学活动非常弱时OC/EC的比值(Lim and Turpin,2002b);④观测期间OC/EC比值的最小值或者利用统计学方法计算得到一定最小百分比下的OC/EC比值作为一次排放比(Lim and Turpin,2002a;Fu et al.,2012).排放清单的方法[方法①]是基于调研的源排放比例和不同源的排放比,综合得到该地区的一次源排放比,但是调研的准确性和源排放的动态特征都决定了该方法无法反映复杂源的空间和时间变化,特别是对于局地源排放复杂的中国地区(Cabada et al.,2004).另外,考虑到我国二次有机气溶胶前体物排放浓度高,一次源和二次源并重的复合大气污染情况,很难区分得到仅有一次源贡献的时间段,因此方法②和③的引入会导致(OC/EC)pri值偏高,并趋向于低估二次源贡献.在实现了元素碳和有机碳在线测量的基础上,方法④是较为常用的估算(OC/EC)pri的方法.其基于观测中有机气溶胶测量得到OC/EC的真实比值,认为一次源中OC/EC的比值属于最小值或者一定最小百分比下的OC/EC比值,该比值基于观测结果在一定程度上更能反映该观测的真实情况.虽然也很难认定最小的(OC/EC)pri的比值就一定没有二次源贡献的影响,却是目前应用较为广泛的(OC/EC)pri的计算方法.Hu等(2012)引进了一种新的OC/EC比值的确定方式.该方式利用数学方法,认为SOC和POC来自于一次和二次来源并且一次源和二次源不相关,将SOC和POC相关性最差时所对应的OC/EC比值作为(OC/EC)pri.这种(OC/EC)pri求解方法减少了人为判断所引起的误差,但其假设SOC和POC来源完全不同,也引入了一定的不确定性(Robinson et al.,2007).
在我国,受到元素碳示踪物法本身的局限性的影响,一次和二次有机气溶胶估算在源排放较为复杂的地区会有一定偏差,但容易被忽略.为了明确元素碳示踪物法应用于源复杂地区的二次有机碳估算的局限性和不确定性,本论文以北京冬季和夏季,及中国东部长岛的有机碳/元素碳观测数据为例,利用元素碳示踪物法,分别计算三次观测得到的二次有机碳浓度及其在有机碳中的比例,并与基于气溶胶质谱的正定矩阵因子(positive matrix factorization,PMF)的源解析结果进行比对.对元素示踪物法结果的不确定性进行讨论和评价,为后续的研究工作提供参考.
2 采样和分析(Samples and analysis) 2.1 采样地点北京观测地点位于北京市邻近北四环的北京大学校内(北纬 39.99,东经116.31).该站点是能够代表北京典型城市大气环境的长期观测站点之一.观测平台设在北京大学1号理科教学楼六楼楼顶,距离地面大约有20 m.仪器采样切割头离楼顶约1.5 m,楼顶用地砖铺砌,不易起尘.观测站点距离南边的四环主路大约500 m,距离东边的中关村北路大约200 m,西面和北面是校园,无明显局地源影响.北京观测的采样时间分别为2010年冬季(11月22日—12月22日)和2011年夏季(8月3日—9月15日).
长岛观测站点位于渤海湾长岛县(北纬37°59′19″,东经120°41′45″).长岛在胶东和辽东半岛之间,无局地重工业影响.受上风向的京津唐地区以及南部的山东半岛区域传输影响,长岛地区大气颗粒物人为源影响强烈.观测站点具体位置位于长岛最北部的一个山顶,距离地面约50 m,周围无高山阻挡.东、北和西三面临海,南面以耕地为主,山顶周围有公路.长岛观测的采样时间为3月21日—4月24日.
2.2 采样仪器本研究中有机碳和元素碳的数据分别来自于(1) 高分辨率飞行时间气溶胶质谱(High-resolution time-of-flight aerosol mass spectrometer,HR-ToF-AMS)和(2)5012 型多角度光散射碳黑气溶胶分析仪器(Multi-angle absorption photometer,MAAP)/黑碳仪(Aethalometer).AMS可以测量亚微米颗粒物中的主要化学组成的浓度及其粒径分布.该仪器能测量的颗粒物主要化学组成包括:有机气溶胶(OA),硫酸盐,硝酸盐,铵盐和氯离子.AMS的具体工作和操作原理介绍请见Canagaratna等(2007).此研究中AMS的测量时间分辨率为4 min.为了定量颗粒物浓度,AMS每隔5~7 d便通入已知浓度的400 nm硝酸铵颗粒物来标定仪器.具体操作和结果请详见Hu等(2013).AMS直接测量可获得OA的浓度.利用AMS测量得到的OA浓度可转化为OC浓度.利用此OC浓度带入元素碳示踪物法计算SOC浓度,可与基于AMS测量得到的有机气溶胶的源解析结果进行比较,深入综合了解判断元素碳示踪物法的局限性.而AMS中OC浓度的获得是通过对AMS测量得到的高质量分辨率有机气溶胶质谱进行元素构成(碳 C、 氢 H、 氧 O和氮 N等)分析,得到有机气溶胶中OA/OC的比值,如公式(2) (Aiken et al.,2007;Aiken et al.,2008)所示:
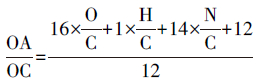
|
(2) |
AMS每4 min测量1次有机气溶胶的谱图,经元素分析可以得到相应OA/OC的值,进而得到OA/OC的时间序列.AMS分析得到的OA/OC的值已经过实验室标定矫正.该方法具体信息请详见其他相关文献(Aiken et al.,2007;Canagaratna et al.,2015).
为配合AMS 高时间分辨率的数据,本研究将利用MAAP和Aethalometer光学法测量得到的黑碳(BC)数据代替元素碳示踪物法中的元素碳(EC)数据.黑碳和元素碳在定义上主要通过其测量方法区分.黑碳主要通过光学仪器测量而元素碳是通过热学仪器测量(Seinfeld and Pandis,1998).外场比对结果显示黑碳和元素碳测量的时间序列通常具有较好的一致性(r > 0.8),这里所用的黑碳的测量方法(MAAP和Aethalometer)与在线光热法测量的元素碳(NOISH方法)测量的绝对浓度的不确定性通常在50%以内(Hitzenberger et al.,2006;Reisinger et al.,2008;兰紫娟 et al.,2011).本文中MAAP和Aethalometer的时间分辨率分别为5 min和2 min.因此,本文中OC/EC比值本质为OC/BC,但为与元素碳示踪物法的习惯用法相统一,后续仍采用OC/EC代称.
3 结果与讨论(Results and discussion) 3.1 一次源(OC/EC)pri的求解本研究选择应用引言中介绍的方法④,基于观测结果求解(OC/EC)pri比值.以观测得到的OC/EC数据为一个整体,研究不同OC/EC比值百分位下所对应的OC和EC散点的回归斜率和相关系数的变化,确定最终(OC/EC)pri.
以北京夏季为例,北京夏季按照OC/EC比值从小到大排列,获得不同百分位(通常选择5%、10%、20%……)下OC/EC比值,然后分别选取该OC/EC比值百分位下所对应的OC和EC的散点,进行线性回归,得到对应的回归斜率和相关系数.为减少截距所带来的不确定性,强制截距为0.如不强制截距为0,回归得到的截距通常为OC最高值的5%.值得注意的是,北京交通政策规定仅夜晚10点钟之后允许大卡车进城,卡车以柴油机为主,柴油机排放的OC/EC比值较汽油机低(Streets et al.,2003),得到的北京夏季白天和夜晚的OC/EC比值分布有所差异(Lin et al.,2009);再加上夜晚没有白天强烈的光化学反应过程,因此将OC/EC的数据以白天和夜晚,分别求解(OC/EC)pri,结果如表 1所示.随着OC/EC比值的百分位数增加,所对应的OC/EC的回归斜率逐渐增加(白天OC/EC斜率在1.6~3.6之间,而夜晚为1.7~3.6).本文选取当OC/EC比值百分位在10%(白天)和5%(夜晚)时,所对应的OC/EC散点的斜率1.79和1.69为白天和夜晚的(OC/EC)pri.该比值比机动车为主要排放源的城市地区OC/EC的比值(2.0)略低(Turpin and Huntzicker,1995;Lim et al.,2003;Cabada et al.,2004),比Lin et al.(2009)所取的(OC/EC)pri(1~1.5)略高.同时,在该(OC/EC)pri下,OC和EC的相关性也最高(r=0.98).
| 表 1 北京夏季不同OC/EC比值的百分位下,所对应的OC和EC回归斜率和相关系数 Table 1 Regression slopes and correlation coefficients between OC and EC at different percentile of OC/EC ratios |
北京冬季和长岛观测(OC/EC)pri的求解过程和北京夏季相类似.但需要指出的是,北京冬季观测发现11月22日—12月11日和12月14日—12月23日两段观测时间,OC和EC受到气象条件和排放源的影响(后段属雾霾静稳天气,有机气溶胶浓度较高),OC/EC比值差别较大,因此对两段时间内的(OC/EC)pri分别进行求解,最终取OC/EC百分位等于30%时,所对应的OC/EC散点的斜率(1.73和2.28)为两段时间的(OC/EC)pri.长岛颗粒物观测浓度无明显日变化和时段差异,则取OC/EC百分位等于10%时,OC/EC散点的斜率值(1.41)为整个观测的(OC/EC)pri.
3.2 比较元素碳示踪物解析和PMF源解析法得到的SOC最终计算得到的3次观测期间SOC的时间序列如图 1所示.作为对比,图中同时展示了PMF源解析方法得到的SOC的时间序列.PMF源解析方法得到SOC是通过解析得到的二次有机气溶胶浓度除以相应因子中OA/OC的比值得到的.3次观测中PMF解析得到的二次有机气溶胶因子和二次无机盐类硫酸盐和硝酸盐都展示了较好的相关性(r > 0.8)(Hu et al.,2013;Hu et al.,2016).北京夏季除二次有机气溶胶外,餐饮源和机动车源分别占总有机气溶胶的21%和13%.而餐饮源(19%)、生物质燃烧源(12%)、机动车源(14%)和煤燃烧源(24%)是北京冬季一次有机气溶胶的主要来源(Hu et al.,2016).长岛地区一次有机气溶胶主要由机动车源(23%)和煤燃烧源(9%)构成(Hu et al.,2013).其中与此后讨论相关的餐饮源有机气溶胶因子的识别是通过:①该因子日变化中规律的中午(12:00—13:00)及傍晚(19:00—21:00)浓度排放高峰(图 3a和b).此排放时间和人们的用餐时间相一致.②其质谱图和其他实验室以及外场观测中餐饮源有机气溶胶质谱图特征一致(He et al.,2010;Mohr et al.,2012).北京冬季生物质燃烧有机气溶胶的判断是通过: ① 该因子的时间序列和生物质燃烧示踪物乙腈的时间序列相一致;②其质谱图中有明显的生物质燃烧的特征离子(m/z 60和m/z 73等)的贡献(Cubison et al.,2011).3次观测中每个因子识别的具体信息以及相应因子特征请参考 Hu等(2013;2015).
 |
| 图 1 北京夏季(a)、北京冬季(b)和长岛地区(c)基于元素碳示踪物法和PMF方法解析得到的SOC的时间序列 Fig. 1 Time series of SOC from EC tracer method and PMF method in summer (a) and winter (b) of Beijing and Changdao island (c) |
 |
| 图 3 北京夏季(a),北京冬季(b)和长岛地区(c)基于元素碳示踪物法和PMF方法计算得到的SOC的日变化图.灰色阴影部分为利用元素碳示踪物法计算得到的SOC的误差 Fig. 3 Diurnal variations of SOC mass concentrations from EC tracer method and PMF method in summer (a) and winter (b) of Beijing and Changdao island (c). The grey area in these plots are the uncertainties (standard deviation) of SOC |
北京夏季观测中,元素碳示踪物法和PMF解析得到的SOC时间序列展现了较为一致的变化趋势.但元素碳示踪物法解析得到的SOC的时间序列的变化范围更大(图 1a).类似的现象同时出现在北京冬季和长岛观测的SOC时间序列比对中(图 1b和c).利用两种方法得到的不同观测地点的SOC平均浓度水平如表 2所示.两种方法计算的SOC浓度差异约为10%~40%.北京夏季和长岛地区PMF源解析方法得到的SOC浓度比用元素碳示踪物法得到的SOC浓度高,与其他研究认为元素碳示踪物法低估了SOC浓度的研究结果相一致(Zhang et al.,2005;Docherty et al.,2008).但是北京冬季元素碳示踪物法得到的SOC浓度却比PMF方法要高.下面对不同观测中造成两种方法估算SOC浓度差异的原因进行分析.
| 表 2 不同地区两种源解析方法得到的SOC、OC和OM/OC的平均浓度水平 Table 2 OC concentration OM/OC ratio and mass concentrations of SOC estimated from EC tracer method and PMF method in three studies |
图 2a中展示了北京夏季元素碳示踪物法和PMF源解析方法计算得到的SOC的散点图.结果显示元素碳示踪物法得到的SOC浓度较PMF方法含有较多的零点,这主要是因为尽管所取的(OC/EC)pri比值已经较低(10%和5%),但是OC/EC散点中仍然存在着OC/EC比值比所取(OC/EC)pri低的点.当OC < EC×(OC/EC)pri,即POC > OC时,设定计算得到的负值SOC浓度为0 μg·m-3.其次,元素碳示踪物法得到的SOC在图 2a中1:1线的右侧有较大偏差的高值.PMF对北京夏季的源解析结果表明北京夏季一次源排放以机动车源(13%)和餐饮源(19%)为主(Hu et al.,2016).利用PMF源解析得到餐饮源有机气溶胶(cooking organic aerosol,COA)占总有机气溶胶的比例进行颜色分类发现,这些1:1线右侧的偏离较大的散点主要受到了餐饮源的影响,随着餐饮源有机气溶胶比例的增加,散点偏差更大.与机动车源不同,餐饮源排放黑碳较少,餐饮源排放的OC/EC(~35)(Hayes et al.,2013)远远高于机动车源(0.57~1.2)(Streets et al.,2003;Shen et al.,2010;Timonen et al.,2014),与二次有机碳中OC/EC比值(> 2)相当(Lim and Turpin,2002b).因此在计算SOC时,会将餐饮源OC归于估算的SOC中.同样类似的现象也在北京冬季元素碳示踪物法与PMF估算的SOC散点的比较中出现.如图 2b所示.北京冬季一次有机气溶胶排放源主要包括机动车、煤燃烧、生物质燃烧和餐饮源.餐饮源和生物质燃烧源中OC/EC比值分别为35和4~150,较其在煤燃烧源(1.1~2.2)和机动车源(0.57~1.2)高(Streets et al.,2003;Shen et al.,2010;Timonen et al.,2014),与二次有机碳中OC/EC比值(> 2)相当.因此北京冬季两方法估算得到的SOC散点的差异原因主要是受到了餐饮源和生物质燃烧的影响.图 2b中展示了利用生物质燃烧源对散点进行颜色分类,当生物质燃烧源占总有机气溶胶比例较高时,1:1线右边散点偏离越大.存在具有较高的(OC/EC)pri一次排放源是造成北京冬夏两季两种方法差异的重要原因.长岛地区两种方法估算的SOC则展现了较好的一致性,相关系数r高达0.84,并且煤燃烧有机气溶胶的比例并没有对SOC的估算造成很大的偏差.这主要是由于长岛地区一次有机气溶胶主要由于(1)煤燃烧和机动车源组成,两者OC/EC排放比相类似;(2)长岛地区燃煤煤燃烧有机碳的比例较低(~9%).长岛地区PMF估算的SOC比元素碳示踪物法估算的SOC高26%,这可能主要由于长岛地区(OC/EC)pri受到大气二次生成的影响仍比实际(OC/EC)pri高的原因.长岛地区元素碳示踪物法相较其他源解析方发低估SOC与其他观测相一致(Zhang et al.,2005).
 |
| 图 2 北京夏季(a,b)、北京冬季(c)和长岛地区(d)基于元素碳示踪物法和PMF方法解析得到的SOC的散点图 其中(a)中利用北京夏季餐饮源有机气溶胶(cooking organic aerosol, COA)占总有机气溶胶比例对散点进行颜色分类.(b)中元素碳示踪物法估算的SOC是在总有机碳中将餐饮源有机碳扣除后,估算的SOC.(c)利用北京冬季生物质燃烧有机气溶胶(biomass burning organic aerosol, BBOA)占总有机气溶胶比例进行颜色分类.(d)中散点利用煤燃烧有机气溶胶(coal combustion organic aerosol, CCOA)在有机气溶胶中比例进行颜色分类 Fig. 2 Scatter plots of SOC from EC tracer method and PMF method in summer (a, b) and winter of Beijing (c) and Changdao island (d). Scatters in the Fig 2(a)was color-coded by the fraction of cooking OA(COA)in total OA. SOC estimated by EC tracer method in Fig 2(b)is done by OC minus cooking OC. Scatters in the Fig 2(c)was color-coded by the fraction of biomass burning OA(BBOA)in total OA. Scatters in the Fig 2(d)was color-coded by the fraction of coal combustion OA(CCOA)in total OA |
为了进一步证明北京冬夏季元素碳示踪物法计算得到的SOC受到了一次源(餐饮源和生物质燃烧)的影响,3次观测中,利用元素碳示踪物法计算得到的SOC和PMF源解析得到的SOC的日变化如图 3所示.北京夏季利用元素碳示踪物法计算得到的SOC具有明显的中午和傍晚的日高峰,与利用PMF解析得到的餐饮源有机气溶胶排放时间相一致.如前文所述,餐饮源的OC/EC排放比较高(~35),利用元素碳示踪物法估算时,很难将其与SOC区别,容易将其归于二次源,造成偏差.元素碳示踪物法解析得到的SOC日变化具有餐饮源有机气溶胶独有的特征,与人们的用餐习惯相一致,进一步证明了北京夏季利用元素碳示踪物法计算得到的SOC在一定程度上受到了餐饮源排放的干扰.Lin et al.(2009)利用元素碳示踪物法对2006年北京夏季在线OC/EC分析仪测量得到的OC进行一次和二次来源估算,其SOC的日变化同样显示了明显的中午和傍晚的高峰,与图 3中餐饮源有机碳出峰时间一致,表明了该研究解析得到的SOC也受到了餐饮源的影响.利用元素碳示踪物法解析得到的北京冬季SOC的日变化也在中午和傍晚相类似时间有所增加,表明与北京夏季估算结果相类似,北京冬季元素碳示踪物法估算得到的SOC也受到了餐饮源排放的影响.这与北京冬季PMF源解析中19%的有机气溶胶来自餐饮源排放相一致.与PMF解析得到的北京冬季SOC相反,元素碳示踪物法解析得到的北京冬季SOC显示了夜间高,白天低的特点,夜间SOC的浓度约是PMF解析得到的SOC浓度的2~3倍,这与北京冬季根据气溶胶质谱结果解析得到的生物质燃烧有机气溶胶浓度的日变化相类似,如图 3b所示.在此观测中,生物质燃烧有机气溶胶的夜间(晚18点—次日早上7点)平均浓度为5 μg·m-3,也是其日间(早7点—晚18点)浓度2 μg·m-3的2~3倍.而长岛地区,元素碳示踪物法和PMF解析得到的SOC的日变化趋势相似,与图 2c中散点较高的线性相关相一致.
3.4 北京夏季剔除餐饮源有机碳后的SOC估算为了进一步探讨具有较高(OC/EC)pri的一次排放源对元素碳示踪物法估算二次有机碳的影响,北京夏季观测为例,将观测期间的餐饮源有机碳从总有机碳数据中扣除,应用元素碳示踪物法对二次有机碳的含量进行了重新估算.餐饮源有机碳的剔除基于AMS测量的有机气溶胶的PMF源解析结果.
元素碳示踪物法的估算方法与前文介绍一致.计算不同百分比下(OC/EC)pri的值,如表 1所示.选取OC/EC比值百分位在10%(白天)和5%(夜晚)时,对应的OC/EC比值为(OC/EC)pri(分别为1.48和1.12),最后计算得到的SOC时间序列,如图 1a所示.图 1a展示了元素碳示踪物法计算得到的SOC与PMF源解析得到的SOC时间序列变化较为一致.图 2b中展示元素碳示踪物法和PMF源解析得到的SOC的散点图,其相关系数r为0.92,远高于包括餐饮有机碳时的元素碳示踪物法计算的SOC和PMF解析得到的SOC的相关性0.56(图 2a).在剔除餐饮源后,元素碳示踪物法估算的SOC的日变化与PMF源解析方法也表现了较好的一致性,都表现为明显的中午出峰,与之前元素碳示踪物法在包括餐饮源时估算的SOC中午和傍晚出峰显著不同(图 3a).剔除餐饮源后,元素碳示踪物法估算的SOC浓度为(5.7±4.1)μg·m-3,比之前包括餐饮源估算的SOC((7.6±5.7)μg·m-3)降低了25%,表明餐饮源对元素碳示踪物法估算的SOC会造成较大偏差.虽然在剔除餐饮源后元素碳示踪物法估算的SOC和PMF源解析得到的SOC展示了较高的一致性,但元素碳示踪物法估算的SOC的平均值((5.7±4.1)μg·m-3)比PMF源解析的SOC结果(8.7 μg·m-3)也低了34%.元素碳示踪物法估算的SOC值较PMF法低,可能是由于(OC/EC)pri值较高所致.北京夏季二次生成强烈,很难剔除二次生成对(OC/EC)pri的影响,这与之前其他文献综述两种方法的比较相一致(Zhang et al.,2005;Docherty et al.,2008;Hu et al.,2012).如果我们利用AMS解析得到的机动车源有机碳和黑碳进行回归,得到的(OC/EC)pri比值在0.55,元素碳示踪物法利用该比值计算得到的SOC浓度((8.8±5.2)μg·m-3)与AMS解析得到的SOC浓度(8.7 μg·m-3)相当.两方法的线性回归的斜率约为1,如图 2b所示.但元素碳示踪物法中,仅利用OC/EC测量值得到的(OC/EC)pri即使在取最小OC/EC百分比(5%)下,其值(1.12和1.19)也为机动车源有机碳和黑碳(0.55)比值的两倍.表明在二次生成强烈的地区(如北京夏季),仅利用OC/EC的测量值,(OC/EC)pri的确容易被高估,从而低估SOC的浓度及其在有机气溶胶中的比例.
4 结论(Conclusions)本研究利用元素碳示踪物法估算了北京夏季、冬季和长岛地区的观测的SOC浓度,其平均浓度分别为(7.6±5.7)μg·m-3,(8.6±14.3)μg·m-3和(3.7±2.8)μg·m-3,与PMF方法解析得到的SOC平均浓度(分别为(8.7±5.2)μg·m-3,(6.0±6.2)μg·m-3和(4.5±3.5)μg·m-3)相差10%~40%.两种方法时间序列和日变化比对结果显示元素碳示踪物法估算的北京夏季SOC明显受到了餐饮源有机碳的影响.北京冬季元素碳示踪物法估算的SOC同时受到了餐饮源和生物质燃烧的影响.因为餐饮源和生物质燃烧源具有较高OC/EC排放比(~35和4~150),远高于煤燃烧源(1.1~2.2)和机动车源(0.57~1.2),与二次有机碳OC/EC比值(> 2)相当.这种具有较高OC/EC比值的一次源容易被归为SOC从而导致SOC估算的误差.因此①在一次源排放较为复杂的情况下,例如中国北京城市地区,如果不同一次源的OC/EC排放比差异过大,特别是具有较高OC/EC排放比的一次源元素碳示踪物法无法准确对复杂源排放地区的大气有机气溶胶进行来源解析.而长岛地区元素碳示踪物法估算得到的SOC和PMF源解析的SOC浓度相当,变化趋势较为一致.②对于一次源类型较为简单的乡村偏远地区,元素碳示踪物法是一个可以简便估算SOC浓度的工具.但是受到方法自身的局限性,OC/EC一次排放比容易受到大气二次生成的影响被高估,从而SOC浓度容易被低估(例如长岛地区低估26%).
建议在利用元素碳示踪物法进行源解析之前,对数据进行综合分析,利用日变化或者其他的示踪物种对该地区的有机碳的来源进行预判.例如,生物质燃烧源可以利用其源示踪物(左旋葡聚糖,钾离子,或乙腈等)进行预判(Andreae and Merlet,2001;Yuan et al.,2010),剔除生物质燃烧影响的时间段后再利用元素碳示踪物法进行估算.而该地区是否有强烈的餐饮源则可以通过有机气溶胶日变化是否在中午和傍晚有明显高峰进行判断(Huang et al.,2010).对于有时间排放规律的一次源,还可以按照时间排放规律进行一次源OC/EC排放比的估算.在无法剔该类一次源影响的情况下,应该考虑到估算的不确定性.
| [1] | Aiken A C, DeCarlo P F, Jimenez J L. 2007.Elemental analysis of organic species with electron ionization high-resolution mass spectrometry[J]. Anal Chem, 79 (29): 8350–8358. |
| [2] | Aiken A C, Decarlo P F, Kroll J H, et al. 2008.O/C and OM/OC ratios of primary, secondary, and ambient organic aerosols with high-resolution time-of-flight aerosol mass spectrometry[J]. Environ Sci Technol, 42 (12): 4478–4485. |
| [3] | Andreae M O, Merlet P. 2001.Emission of trace gases and aerosols from biomass burning[J]. Global Biogeochem Cy, 15 (4): 955–966. |
| [4] | Cabada J C, Pandis S N, Subramanian R, et al. 2004. Estimating the secondary organic aerosol contribution to PM2.5 using the EC tracer method[J]. Aerosol Sci Tech, 38(12):140-155 |
| [5] | Canagaratna M R, Jayne J T, Jimenez J L, et al. 2007.Chemical and microphysical characterization of ambient aerosols with the aerodyne aerosol mass spectrometer[J]. Mass Spectrom Rev, 26 (2): 185–222. |
| [6] | Canagaratna M R, Jimenez J L, Kroll J H, et al. 2015.Elemental ratio measurements of organic compounds using aerosol mass spectrometry:characterization, improved calibration, and implications[J]. Atmos Chem Phys, 15 (1): 253–272. |
| [7] | Cao J J, Lee S C, Ho K F, et al. 2004.Spatial and seasonal variations of atmospheric organic carbon and elemental carbon in Pearl River Delta Region, China[J]. Atmos Environ, 38 (27): 4447–4456. |
| [8] | Cao J J, Lee S C, Chow J C, et al. 2007.Spatial and seasonal distributions of carbonaceous aerosols over China[J]. J Geophys Res-Atmos, 112 : D22S11. |
| [9] | Chu S H. 2005.Stable estimate of primary OC/EC ratios in the EC tracer method[J]. Atmos Environ, 39 (8): 1383–1392. |
| [10] | Cubison M J, Ortega A M, Hayes P L, et al. 2011.Effects of aging on organic aerosol from open biomass burning smoke in aircraft and laboratory studies[J]. Atmos Chem Phys, 11 (23): 12049–12064. |
| [11] | Docherty K S, Stone E A, Ulbrich I M, et al. 2008.Apportionment of Primary and Secondary Organic Aerosols in Southern California during the 2005 Study of Organic Aerosols in Riverside (SOAR-1)[J]. Environ Sci Technol, 42 (20): 7655–7662. |
| [12] | Duan J C, Tan J H, Cheng D X, et al. 2007.Sources and characteristics of carbonaceous aerosol in two largest cities in Pearl River Delta Region, China[J]. Atmos Environ, 41 (14): 2895–2903. |
| [13] | Feng J L, Guo Z G, Zhang T R, et al. 2012.Source and formation of secondary particulate matter in PM2.5 in Asian continental outflow[J]. J Geophys Res, 117 : D03302. |
| [14] | Fu T M, Cao J J, Zhang X Y, et al. 2012.Carbonaceous aerosols in China:top-down constraints on primary sources and estimation of secondary contribution[J]. Atmos Chem Phys, 12 (5): 2725–2746. |
| [15] | Hayes P L, Ortega A M, Cubison M J, et al. 2013.Organic aerosol composition and sources in Pasadena, California, during the 2010 CalNex campaign[J]. J Geophys Res, 118 (16): 9233–9257. |
| [16] | He L Y, Lin Y, Huang X F, et al. 2010.Characterization of high-resolution aerosol mass spectra of primary organic aerosol emissions from Chinese cooking and biomass burning[J]. Atmos Chem Phys, 10 (23): 11535–11543. |
| [17] | Hitzenberger R, Petzold A, Bauer H, et al. 2006.Intercomparison of thermal and optical measurement methods for elemental carbon and black carbon at an urban location[J]. Environ Sci Technol, 40 (20): 6377–6383. |
| [18] | Hu W W, Hu M, Hu W, et al. 2016.Chemical composition, sources and aging process of sub-micron aerosols in Beijing:contrast between summer and winter[J]. J Geophys Res, 121 (4): 1955–1977. |
| [19] | Hu W W, Hu M, Deng Z Q, et al. 2012.The characteristics and origins of carbonaceous aerosol at a rural site of PRD in summer of 2006[J]. Atmos Chem Phys, 12 (4): 1811–1822. |
| [20] | Hu W W, Hu M, Yuan B, et al. 2013.Insights on organic aerosol aging and the influence of coal combustion at a regional receptor site of central eastern China[J]. Atmos Chem Phys, 13 (19): 10095–10112. |
| [21] | Huang X F, He L Y, Hu M, et al. 2010.Highly time-resolved chemical characterization of atmospheric submicron particles during 2008 Beijing Olympic Games using an Aerodyne High-Resolution Aerosol Mass Spectrometer[J]. Atmos Chem Phys, 10 (18): 8933–8945. |
| [22] | 兰紫娟, 黄晓峰, 何凌燕, 等.2011.不同碳质气溶胶在线监测技术的实测比较研究[J].北京大学学报(自然科学版), vol.47, vol.47 :159–165. |
| [23] | Lim H J, Turpin B J. 2002.Origins of Primary and Secondary Organic Aerosol in Atlanta:Results of time-resolved measurements during the Atlanta Supersite experiment[J]. Environ Sci Technol, 36 (21): 4489–4496. |
| [24] | Lim H J, Turpin B J, Russell L M, et al. 2003.Organic and elemental carbon measurements during ACE-Asia suggest a longer atmospheric lifetime for elemental carbon[J]. Environ Sci Technol, 37 (14): 3055–3061. |
| [25] | Lin P, Hu M, Deng Z, et al. 2009.Seasonal and diurnal variations of organic carbon in PM2[J]. 5 in Beijing and the estimation of secondary organic carbon[J]. J Geophys Res, 114 : . |
| [26] | Mohr C, DeCarlo P F, Heringa M F, et al. 2012.Identification and quantification of organic aerosol from cooking and other sources in Barcelona using aerosol mass spectrometer data[J]. Atmos Chem Phys, 12 (4): 1649–1665. |
| [27] | Reisinger P, Wonaschutz A, Hitzenberger R, et al. 2008.Intercomparison of measurement techniques for black or elemental carbon under urban background conditions in wintertime:Influence of biomass combustion[J]. Environ Sci Technol, 42 (3): 884–889. |
| [28] | Robinson A L, Donahue N M, Shrivastava M K, et al. 2007.Rethinking organic aerosols:Semivolatile emissions and photochemical aging[J]. Science, 315 : 1259–1262. |
| [29] | Seinfeld J H, Pandis S N. 1998. Atmospheric Chemistry and Physics:from Air Pollution to Climate Change[B]. John Wiley, New York |
| [30] | Shen G, Yang Y, Wang W, et al. 2010.Emission Factors of Particulate Matter and Elemental Carbon for Crop Residues and Coals Burned in Typical Household Stoves in China[J]. Environ Sci Technol, 44 (18): 7157–7162. |
| [31] | Streets D G, Bond T C, Carmichael G R, et al. 2003. An inventory of gaseous and primary aerosol emissions in Asia in the year 2000. J Geophys Res[M]. . |
| [32] | Timonen H, Jaffe D A, Wigder N, et al. 2014.Sources of carbonaceous aerosol in the free troposphere[J]. Atmos Environ, 92 (0): 146–153. |
| [33] | Turpin B J, Huntzicker J J. 1991.Secondary Formation of Organic Aerosol in the Los-Angeles Basin-a Descriptive Analysis of Organic and Elemental Carbon Concentrations[J]. Atmos Environ a-Gen, 25 (2): 207–215. |
| [34] | Turpin B J, Huntzicker J J. 1995.Identification of Secondary Organic Aerosol Episodes and Quantitation of Primary and Secondary Organic Aerosol Concentrations during Scaqs[J]. Atmos Environ, 29 (23): 3527–3544. |
| [35] | Xiao R, Takegawa N, Zheng M, et al. 2011.Characterization and source apportionment of submicron aerosol with aerosol mass spectrometer during the PRIDE-PRD 2006 campaign[J]. Atmos Chem Phys, 11 (14): 6911–6929. |
| [36] | Yuan B, Liu Y, Shao M, et al. 2010.Biomass Burning Contributions to Ambient VOCs Species at a Receptor Site in the Pearl River Delta (PRD), China[J]. Environ Sci Technol, 44 (12): 4577–4582. |
| [37] | Zhang Q, Worsnop D R, Canagaratna M R, et al. 2005.Hydrocarbon-like and oxygenated organic aerosols in Pittsburgh:insights into sources and processes of organic aerosols[J]. Atmos Chem Phys, 5 : 3289–3311. |