 2017, Vol. 38
2017, Vol. 38



2. 第二军医大学长海医院脊柱外科, 上海 200433
2. Department of Spine Surgery, Changhai Hospital, Second Military Medical University, Shanghai 200433, China
多发性骨髓瘤(multiple myeloma,MM)是骨髓浆细胞克隆性增殖的血液系统恶性肿瘤[1],约占全身恶性肿瘤的1%,占所有血液系统肿瘤的10%。MM患者常伴有骨骼病变、高钙血症、严重的免疫缺陷以及细菌感染[2]。在我国,MM是第二常见的血液恶性肿瘤[3],主要通过化疗、造血干/祖细胞移植、药物治疗和免疫治疗等方式进行治疗[4],但术后5年生存率仅为44.9%[5]。目前,已有许多研究开始探索预防和治疗MM的新方法。B淋巴细胞刺激因子可作为诊断和治疗MM的生物标志物[6];血清中自由轻链比例升高可作为无症状性MM进展的预测指标[7];而在药物治疗方面,选择性蛋白酶抑制剂硼替佐米也表现出良好的治疗效果[8]。Lund等[9]报道沙利度胺联合美法仑+泼尼松治疗可延长MM患者的生存时间,但该治疗方式可能会引起疲劳、皮疹、多发性神经病和静脉血栓栓塞等并发症[10]。由于MM具有异质性较强、老年患者多见、对化疗药物的反应各异等特点,仍需进一步寻找MM早期特异性的诊断标志物及有效的治疗方法,延长患者的生存时间和提高其生活质量。
高通量基因芯片在肿瘤的分子诊断、肿瘤分类、患者预后和预测分析等方面应用广泛,有望为研究MM的发病机制和分子诊断提供新途径。本研究运用生物信息学技术对MM患者和健康对照组人群的基因表达芯片数据进行分析,挖掘MM患者和健康对照组人群的差异表达基因(differentially expressed genes,DEGs),并通过基因富集分析和通路分析、蛋白相互作用网路的构建以及蛋白网络的模块分析,探索MM相关基因通路和功能的变化,旨在为MM的早期诊断和基因治疗提供新的研究思路和理论依据。
1 材料和方法 1.1 基因表达芯片数据获取由于MM的发病率相对其他肿瘤较低,肿瘤基因组图谱(the cancer genome atlas,TCGA)数据库(https://cancergenome.nih.gov/)中未找到相关芯片样本集,故本研究的芯片数据集中于GEO数据库(https://www.ncbi.nlm.nih.gov/gds/)。从GEO数据库中根据以下3条标准进行芯片筛选:(1) MM患者标本,而非动物模型;(2) 多发性;(3) 有原始的高通量芯片数据。得到André等[11]上传的基于GPL570芯片分析平台(Affymetrix Human Genome U133 Plus 2.0 Array)的GSE36474样本集、López-Corral等[12]上传的基于GPL6244芯片分析平台(Affymetrix Human Gene 1.0 ST Array)的GSE47552样本集、Gutiérrez等[13]上传的基于GPL96芯片分析平台(Affymetrix Human Genome U133A Array)的GSE6691样本集共3个不同的芯片样本集。GSE36474芯片样本细胞种类为间充质干细胞,样本量为7;GSE47552芯片样本细胞种类为浆细胞,样本量为46;GSE6691芯片样本细胞种类为浆细胞,样本量为21。
1.2 样本均衡性检验和DEGs的筛选分别对3个样本集进行均衡性检验,主要包括各个样本集MM组和对照组的性别、年龄等。3个样本集的原始文件被用于DEGs的筛选。将芯片样本集分别导入Morpheus在线工具(https://software.broadinstitute.org/morpheus/)进行数据质量控制。质量控制时要求:(1) 基因中位数值至少发生2倍改变;(2) 基因表达量变异P值小于0.05;(3) 数据缺失值不超过50%。基因过滤后,根据层次聚类将数据分为两组:有相似表达模式的MM组和健康对照组。对MM组和健康对照组行t检验,满足P<0.05且倍数变化>2或 < 0.5的为DEGs,其中倍数变化 < 0.5为表达下调DEGs,倍数变化>2为表达上调DEGs。
1.3 DEGs的功能分析和基因富集分析通过DAVID数据库(https://david.ncifcrf.gov/)在基因功能层面对DEGs进行基因本体(gene ontology,GO)富集分析[14]和京都基因与基因组百科全书(Kyoto encyclopedia of genes and genomes,KEGG)分析[15]。将上调或下调的DEGs分别输入DAVID数据库中,并将基因名称转化为Official-Gene-Symbol,选取GO富集结果中的GOTERM_BP_DIRECT(生物过程)、GOTERM_MF_DIRECT(分子功能)和GOTERM_CC_DIRECT(细胞成分)进行GO富集分析。此外,选取通路结果中的KEGG_PATHWAY,对DEGs的集中信号通路进行分析。
1.4 蛋白质相互作用网络的建立和模块分析为进一步挖掘DEGs中的核心基因,研究通过STRING数据库[16](http://string-db.org/)在线分析DEGs对应编码蛋白的相互作用,筛选有实验验证的、综合得分为0.4以上(满分为1) 的基因构建相互作用网络。使用信息分析学软件Cytoscape(3.4.0版本)[17]分析STRING数据库构建出的蛋白质相互作用网络,通过Cytoscape的MCODE功能得出蛋白质相互作用网络中的相关模块,标准设置如下:(1) MCODE得分>3;(2) 节点数>4.0。分析每个相关模块的功能和通路丰度。
1.5 统计学处理MM组和健康对照组的均衡性检验和DEGs筛选通过Morpheus在线工具自带统计学工具行χ2检验和t检验。GO分析通过DAVID数据库自带统计学工具使用弗朗尼校正法(Bonferroni),本杰明假阳性率法(Benjamini false discovery rate)和靴带法(boot straping)等方法进行检验。KEGG分析通过DAVID数据库自带统计学工具进行Fisher精确概率检验及基因集富集分析(gene set enrichment analysis)。蛋白质相互作用网络通过Cytoscape软件进行Pearson相关系数分析后,将基因表达数据进行层次聚类,连接方法为平均连接聚类法(average linkage)和中位数标准化。检验水准(α)为0.05。
2 结果 2.1 样本均衡性检验和DEGs的筛选3个样本集中MM组与健康对照组的性别、年龄等差异均无统计学意义(P>0.05),共对基因表达芯片数据中的61例MM患者和13名健康对照组受试者的数据进行了分析。获得16 211个DEGs,其中表达上调的基因有7 586个,下调的有8 625个。DEGs热图(前50位上调基因和前50位下调基因,以GSE36474样本集为例)见图 1。

|
图 1 GSE36474样本集中DEGs的热图 Fig 1 Heat map of differentially expressed genes (DEGs)in sample set GSE36474 The top 50 up-regulated genes and the top 50 down-regulated genes. Red: Up-regulation; Blue: Down-regulation. MM: Multiple myeloma |
2.2 GO功能分析
将所有筛选所得的DEGs通过DAVID数据库进行分析,得出有代表性的GO类别和KEGG通路。GO分析结果显示,生物学过程中上调的DEGs主要涉及鞘糖脂代谢过程(glycosphingolipid metabolic process)、DNA转录过程(transcription, DNA-templated)和干扰素γ介导的信号转导通路(interferon-gamma-mediated signaling pathway)等30个功能簇;下调的DEGs主要涉及细胞分裂(cell division)、DNA复制(DNA replication)和有丝分裂核分裂(mitotic nuclear division)等163个功能簇。关于分子功能,上调的DEGs主要涉及蛋白结合(protein binding)、转录因子与特异序列DNA结合的活性(transcription factor activity, sequence-specific DNA binding)和烟酰胺腺嘌呤二核苷酸结合(NAD binding)等29个功能簇;而下调的DEGs主要涉及组蛋白结合(histone binding)、多聚A尾RNA结合[poly(A) RNA binding]和DNA结合(DNA binding)等59个功能簇。此外,GO细胞成分分析显示,上调的DEGs主要集中在细胞溶质(cytosol)、溶酶体膜(lysosomal membrane)和溶酶体(lysosome)等27个功能簇中;而下调的DEGs主要集中在核质(nucleoplasm)、细胞核(nucleus)和细胞质(cytoplasm)等78个功能簇中。部分GO分析结果见表 1。
|
|
表 1 MM患者中DEGs的GO分析 Tab 1 Gene ontology analysis of differentially expressed genes (DEGs)associated with MM |
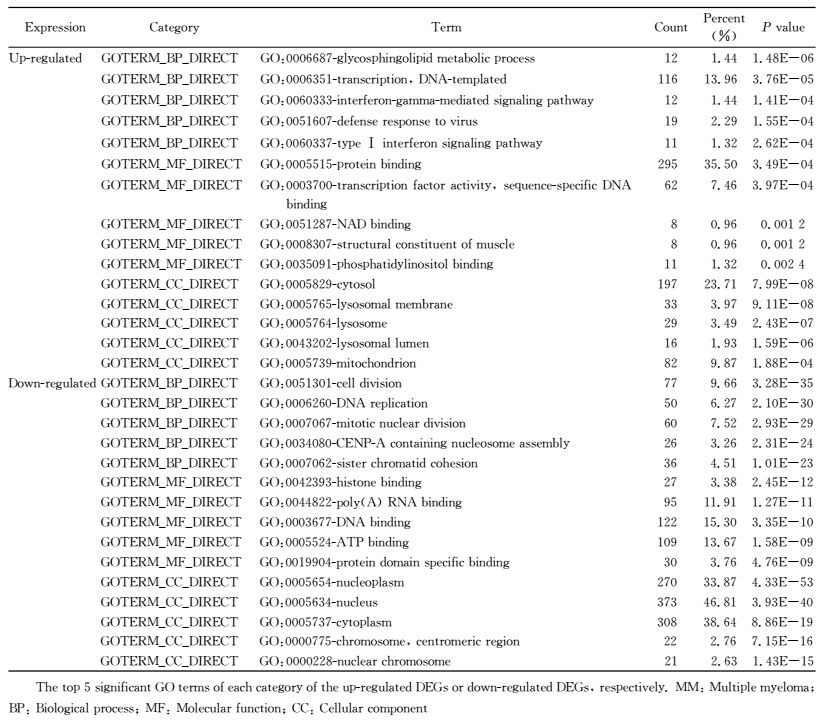
2.3 KEGG信号通路分析
通过KEGG分析富集得到差异最显著的上调和下调DEGs所在的信号通路。上调的DEGs主要涉及溶酶体相关通路(lysosome)、病毒致癌作用(viral carcinogenesis)和丙型肝炎相关通路(hepatitis C)等26条信号通路;而下调的DEGs主要涉及DNA复制(DNA replication)、Fanconi贫血通路(Fanconi anemia pathway)和卵母细胞减数分裂(oocyte meiosis)等27条信号通路(表 2)。
|
|
表 2 MM患者中DEGs的KEGG分析 Tab 2 KEGG pathway analysis of differentially expressed genes (DEGs) associated with MM |

2.4 蛋白质相互作用网络中的模块分析
基于STRING数据库筛选出得分最高的前10位核心基因,分别是细胞周期蛋白依赖性激酶1 (cyclin-dependent kinase 1,CDK1)、拓扑异构酶Ⅱ α亚基[topoisomerase (DNA) Ⅱ alpha,TOP2A]、光激酶B(aurora kinase B,AURKB)、乳腺癌易感基因1 (breast cancer susceptibility gene 1,BRCA1)、细胞周期检测点激酶1 (checkpoint kinase 1,CHEK1)、磷酸酶张力蛋白同源物(phosphatase and tensin homolog,PTEN)、RAD51、单磷酸鸟嘌呤合成酶(guanine monphosphate synthetase,GMPS)、细胞分裂周期45同源物(cell division cycle 45 homolog,CDC45) 和细胞周期蛋白依赖性激酶抑制剂2A (cyclin-dependent kinase inhibitor 2A,CDKN2A)。在这些核心基因中,CDK1的相关节点数最高,为157个。此外,使用Cytoscape的MCODE功能对STRING数据库分析得出的蛋白网络中的1 180个节点蛋白的8 250条关系进行了分析,筛选出MCODE得分最高的3个模块(图 2,表 3)。对3个模块中的基因均进行了功能分析,结果显示,模块1~3中丰度最高的基因主要与核分裂(nuclear division)、DNA复制(DNA replication)和核酸代谢过程(nucleic acid metabolic process)相关。

|
图 2 蛋白质相互作用网络中得分最高的前3个模块 Fig 2 Top 3 modules from the protein-protein interaction network A: Enriched pathways of module 1; B: Enriched pathways of module 2; C: Enriched pathways of module 3 |
|
|
表 3 蛋白质相互作用网络中得分最高的前3个模块及其功能分析 Tab 3 Top 3 modules from the protein-protein interaction network and the related functions |

3 讨论
本研究对GEO数据库中的基因表达芯片数据文件GSE36474、GSE47552和GSE6691中MM患者和健康人的样本信息进行了生物信息学分析,最终筛选出16 211个DEGs,其中7 586个表达上调,8 625个表达下调。与健康对照人群相比,MM患者的样本中上调表达的基因主要有CDK1、TOP2A、AURKB、BRCA1、CHEK1、PTEN、RAD51、GMPS、CDC45和CDKN2A等。
通过GO分析,本研究发现MM患者的上调DEGs主要涉及鞘糖脂代谢过程、DNA转录过程和干扰素γ介导的信号转导通路等生物学过程。鞘糖脂是一类广泛分布在细胞膜上的糖脂类物质,在调控细胞的识别、黏附、增殖和凋亡等生物学过程中有重要作用。Ersek等[18]研究发现,MM中激活破骨细胞需要鞘糖脂的合成,并且激活的破骨细胞能够进一步促进鞘糖脂的合成,形成正反馈作用。在骨髓平衡紊乱疾病的药物治疗中,限制鞘糖脂的合成能够有效预防MM的发生[19]。干扰素γ有着强大的抗肿瘤效果,应用干扰素γ治疗MM疗效明显[20]。Haydaroglu等[21]比较了包括干扰素γ在内的多种不同药物治疗MM的效果,结果发现干扰素γ相关基因及其等位基因可能在MM中发挥作用;本研究在GO分析中也发现MM患者中表达显著上调的基因集中在干扰素γ介导的信号转导通路中。
KEGG通路分析结果表明,DEGs中丰度最高的信号通路是溶酶体相关通路。李礼轩和李佳[22]研究发现,通过慢病毒介导的shRNA沉默溶酶体相关膜蛋白2A的表达可抑制MM细胞增殖。细胞自噬是依赖溶酶体途径对细胞内物质进行处理的重要过程,自噬活性与MM的发生和发展密切相关[23]。KEGG通路分析显示下调的DEGs所在的信号通路主要集中在细胞周期。孙婉玲等[24]分析了MM细胞的DNA含量和细胞周期,发现MM患者处于S+G2/M期的骨髓瘤细胞(CD138+)比例明显上升,这和KEGG的通路富集结果相对应。细胞增殖失调是肿瘤细胞的特点之一,肿瘤细胞通常对调节细胞周期的基因(如CDK1、CHEK1和CDC45等)直接调控,使细胞增殖紊乱。
通过Cytoscape对STRING数据库产生的蛋白相互作用网络进行分析,本研究获得DEGs中得分最高的前10位核心基因,CDK1在真核细胞中通过调节中心体周期以及有丝分裂的发生,对细胞周期的调控起着关键作用,CDK1可以促进G2-M过渡、加速G1过程和促进G1-S过渡[25]。CDK1是细胞进入S期和进行有丝分裂所必需的。BRCA1在乳腺癌发病早期表达上升,并且特异性介导E3泛素蛋白连接酶多聚泛素链中六聚赖氨酸的形成,进而促进细胞对受损DNA的修复。BRCA1-BARD1二聚体协调调控DNA修复、蛋白质泛素化和转录调控等细胞通路,从而维持基因组的稳定性[26]。已有研究报道CHEK1在人类MM中调节细胞G0/G1期[27],Tu等[28]研究发现在MM中抑制CHEK1基因的表达可以缩短S期,促进骨髓瘤细胞凋亡。本研究发现CHEK1表达上调,这和细胞周期密切相关,表明CHEK1可能是治疗MM的一个潜在靶点。
本研究挖掘到了一系列关键基因,但也存在部分局限:(1) 前期缺少相关临床标本,未进行基础实验验证。目前课题组已收集标本,相关实验正在进行中。(2) 数据进行了质量控制,基本信息的均衡性也满足要求,但患者个体间的状态差异未全部提及(GSE36474患者均为MM Ⅲa期,而GSE47552和GSE6691分期未知),提示我们在进行验证实验时需保持患者状态的均一性,减少组内个体差异。(3) 由于TCGA数据库中未收录MM的芯片信息,故研究仅分析了GEO数据库的芯片信息。
综上,本研究通过Morpheus在线工具、DAVID数据库、STRING数据库和Cytoscape等多种生物信息学工具对基因芯片数据进行了筛选、统合、挖掘和分析,探索MM相关分析标签、DEGs、信号通路和功能模块的变化,从多个角度分析MM的表达特征。研究结果表明,鞘糖脂代谢过程和细胞周期可能与MM的发展密切相关,干扰素γ有望成为MM的潜在治疗剂,CDK1、BRCA1-BARD1和CHEK1基因可作为MM治疗的潜在靶点。这些结果为进一步的生物学验证和基础研究提供了依据,也为今后MM的诊断和治疗研究提供了新方向。
| [1] | LONIAL S, ANDERSON K C. Association of response endpoints with survival outcomes in multiple myeloma[J]. Leukemia, 2014, 28: 258–268. DOI: 10.1038/leu.2013.220 |
| [2] | RAAB M S, PODAR K, BREITKREUTZ I, RICHARDSON P G, ANDERSON K C. Multiple myeloma[J]. Lancet, 2009, 374: 324–339. DOI: 10.1016/S0140-6736(09)60221-X |
| [3] | 陈协群. 中国多发性骨髓瘤诊治指南(2015年修订)[J]. 中华内科杂志, 2016, 54: 1066–1070. |
| [4] | 吴玉玲, 黄东平. 多发性骨髓瘤的治疗进展[J]. 现代医药卫生, 2016, 32: 2828–2830. DOI: 10.3969/j.issn.1009-5519.2016.18.015 |
| [5] | WANG X G, PENG Y, SONG X L, LAN J P. Identification potential biomarkers and therapeutic agents in multiple myeloma based on bioinformatics analysis[J]. Eur Rev Med Pharmacol Sci, 2016, 20: 810–817. |
| [6] | JIANG P, YUEGUO W, HUIMING H, HONGXIANG Y, MEI W, JU S. B-lymphocyte stimulator:a new biomarker for multiple myeloma[J]. Eur J Haematol, 2009, 82: 267–276. DOI: 10.1111/ejh.2009.82.issue-4 |
| [7] | LARSEN J, KUMAR S K, DISPENZIERI A, KYLE R, KATZMANN J, RAJKUMAR S V. Serum free light chain ratio as a biomarker for high-risk smoldering multiple myeloma[J]. Leukemia, 2013, 27: 941–946. DOI: 10.1038/leu.2012.296 |
| [8] | RICHARDSON P G, SONNEVELD P, SCHUSTER M W, IRWIN D, STADTMAUER E A, FACON T, et al. Bortezomib or high-dose dexamethasone for relapsed multiple myeloma[J]. N Engl J Med, 2005, 352: 2487–2498. DOI: 10.1056/NEJMoa043445 |
| [9] | LUND J, UTTERVALL K, LIWING J, GAHRTON G, ALICI E, ASCHAN J, et al. Addition of thalidomide to melphalan and prednisone treatment prolongs survival in multiple myeloma——a retrospective population based study of 1162 patients[J]. Eur J Haematol, 2014, 92: 19–25. DOI: 10.1111/ejh.2013.92.issue-1 |
| [10] | HAAS P, DENZ U, IHORST G, ENGELHARDT M. Thalidomide in consecutive multiple myeloma patients:single-center analysis on practical aspects, efficacy, side effects and prognostic factors with lower thalidomide doses[J]. Eur J Haematol, 2008, 80: 303–309. DOI: 10.1111/ejh.2008.80.issue-4 |
| [11] | ANDRÉT, MEULEMAN N, STAMATOPOULOS B, DE BRUYN C, PIETERS K, BRON D, et al. Evidences of early senescence in multiple myeloma bone marrow mesenchymal stromal cells[J]. PLoS One, 2013, 8: e59756. DOI: 10.1371/journal.pone.0059756 |
| [12] | LÓPEZ-CORRAL L, CORCHETE L A, SARASQUETE M E, MATEOS M V, GARCÍA-SANZ R, FERMIÑÁN E, et al. Transcriptome analysis reveals molecular profiles associated with evolving steps of monoclonal gammopathies[J]. Haematologica, 2014, 99: 1365–1372. DOI: 10.3324/haematol.2013.087809 |
| [13] | GUTIÉRREZ N, OCIO E, DE LAS RIVAS J, MAISO P, DELGADO M, FERMINAN E, et al. Gene expression profiling of B lymphocytes and plasma cells from Waldenstr m's macroglobulinemia:comparison with expression patterns of the same cell counterparts from chronic lymphocytic leukemia, multiple myeloma and normal individuals[J]. Leukemia, 2007, 21: 541–549. DOI: 10.1038/sj.leu.2404520 |
| [14] | Gene Ontology Consortium. Gene ontology consortium:going forward[J]. Nucleic Acids Res, 2015, 43: D1049–D1056. DOI: 10.1093/nar/gku1179 |
| [15] | KANEHISA M, GOTO S. KEGG:Kyoto encyclopedia of genes and genomes[J]. Nucleic Acids Res, 2000, 28: 27–30. DOI: 10.1093/nar/28.1.27 |
| [16] | SZKLARCZYK D, FRANCESCHINI A, WYDER S, FORSLUND K, HELLER D, HUERTA-CEPAS J, et al. STRING v10:protein-protein interaction networks, integrated over the tree of life[J]. Nucleic Acids Res, 2015, 43: D447–D452. DOI: 10.1093/nar/gku1003 |
| [17] | SHANNON P, MARKIEL A, OZIER O, BALIGA N S, WANG J T, RAMAGE D, et al. Cytoscape:a software environment for integrated models of biomolecular interaction networks[J]. Genome Res, 2003, 13: 2498–2504. DOI: 10.1101/gr.1239303 |
| [18] | ERSEK A, XU K, ANTONOPOULOS A, BUTTERS T D, SANTO A E, VATTAKUZHI Y, et al. Glycosphingolipid synthesis inhibition limits osteoclast activation and myeloma bone disease[J]. J Clin Invest, 2015, 125: 2279–2292. DOI: 10.1172/JCI59987 |
| [19] | ERSEK A, KARADIMITRIS A, HORWOOD N. Effect of glycosphingolipids on osteoclastogenesis and osteolytic bone diseases[J]. Front Endocrinol (Lausanne), 2012, 3: 106. |
| [20] | 周亚兰, 米瑞华, 艾昊, 陈琳, 袁芳芳, 尹青松, 等. 沙利度胺联合干扰素在复发难治及体能、经济状况欠佳多发性骨髓瘤患者中的应用[J]. 白血病·淋巴瘤, 2016, 25: 479–482. DOI: 10.3760/cma.j.issn.1009-9921.2016.08.008 |
| [21] | HAYDAROGLU H, OGUZKAN BALCI S, PEHLI VAN S, OZDILLI K, GUNDOGAN E, OKAN V, et al. Effect of cytokine genes in the pathogenesis and on the clinical parameters for the treatment of multiple myeloma[J]. Immunol Invest, 2017, 46: 10–21. DOI: 10.1080/08820139.2016.1208219 |
| [22] | 李礼轩, 李佳. 慢病毒介导的shRNA沉默溶酶体相关膜蛋白2A表达可抑制多发性骨髓瘤细胞增殖[J]. 细胞与分子免疫学杂志, 2015, 31: 605–608. |
| [23] | MIZUSHIMA N, KOMATSU M. Autophagy:renovation of cells and tissues[J]. Cell, 2011, 147: 728–741. DOI: 10.1016/j.cell.2011.10.026 |
| [24] | 孙婉玲, 武永吉, 汪玄, 李辉, 庄俊玲. 多发性骨髓瘤细胞DNA含量和细胞周期分析[J]. 中国实验血液学杂志, 2008, 16: 824–828. |
| [25] | BENANTI J A. Create, activate, destroy, repeat:Cdk1 controls proliferation by limiting transcription factor activity[J]. Curr Genet, 2016, 62: 271–276. DOI: 10.1007/s00294-015-0535-5 |
| [26] | MEHRGOU A, AKOUCHEKIAN M. The importance of BRCA1 and BRCA2 genes mutations in breast cancer development[J]. Med J Islam Repub Iran, 2016, 30: 369. |
| [27] | PEI X Y, DAI Y, YOUSSEFIAN L E, CHEN S, BODIE W W, TAKABATAKE Y, et al. Cytokinetically quiescent (G0/G1) human multiple myeloma cells are susceptible to simultaneous inhibition of Chk1 and MEK1/2[J]. Blood, 2011, 118: 5189–5200. DOI: 10.1182/blood-2011-02-339432 |
| [28] | TU Y S, KANG X L, ZHOU J G, LV X F, TANG Y B, GUAN Y Y. Involvement of Chk1-Cdc25A-cyclin A/CDk2 pathway in simvastatin induced S-phase cell cycle arrest and apoptosis in multiple myeloma cells[J]. Eur J Pharmacol, 2011, 670: 356–364. DOI: 10.1016/j.ejphar.2011.09.031 |