 2014, Vol. 35
2014, Vol. 35



2. 三峡大学医学院药理学教研室, 宜昌 443002
2. Department of Pharmacology, Medical Science College, Three Gorges University, Yichang 443002, Hubei, China
肝细胞癌临床上发病率高,中晚期预后不良,且由于外科手术切除不彻底、对放化疗不敏感等原因患者治疗效果并不理想。近年来随着分子生物学的进展和对癌症发病机制的深入研究,出现了以特定分子为靶点的抗肿瘤药物。研究表明表皮生长因子受体(epidermal growth factor receptor,EGFR) 的过表达与肿瘤的恶性程度、浸润转移及不良预后密切相关,是一个重要的分子靶点[ 1,2 ]。目前肝癌分子靶向治疗药物常用EGFR酪氨酸激酶抑制剂(TKI),但EGFR-TKI的使用会导致获得性耐药的发生[ 3 ]。钙黏蛋白(E-cadherin)在多种肿瘤中表达,其表达下降或缺失不仅与肿瘤细胞的低分化、高侵袭性和转移性相关,还直接影响着患者的预后[ 1,2 ]。E-cadherin与EGFR在影响肿瘤的侵袭性、转移及预后方面有着有着密切的联系。本研究拟通过探讨E-cadherin表达在EGFR-TKI治疗肝细胞癌敏感/耐药形成中的作用,对其是否直接参与EGFR-TKI耐药进行初步研究。 1 材料和方法 1.1 材料
上皮性肝癌细胞HepG2、BEL-7404细胞,间质性肝癌细胞MHCC97、SK-HEP-1均购自中国科学院典型培养物保藏委员会细胞库。TRIzol总RNA抽提试剂(Invitrogen公司);RPMI-1640培养液(Gibco公司);青霉素、链霉素(华北制药股份有限公司);小牛血清(杭州四季青生物工程材料有限公司);RT-PCR试剂盒(Fermentas公司);一抗鼠抗人E-cadherin单克隆抗体(Cell Signaling公司),二抗羊抗鼠RAP抗体(武汉谷歌生物科技有限公司);一抗兔抗人β-actin抗体、二抗羊抗兔RAP抗体(武汉谷歌生物科技有限公司);BCA 蛋白定量试剂盒(碧云天生物技术有限公司);转染试剂、G418试剂、MTT试剂(武汉谷歌生物科技有限公司)。EGFR-TKI PD153035、吉非替尼(gefinitib)购自南京德宝生化器材有限公司。E-cadherin质粒由澳大利亚昆士兰大学友情提供。 1.2 MTT法检测细胞增殖
制备单细胞悬液,调整细胞密度为2×105/mL。 加100 μL细胞悬液于96孔培养板,即每孔2×104个细胞,每组3孔。过夜后,翻板倒净培养液,加入含不同浓度药物的培养液处理细胞(PD153035:0、500、1 000、1 500、2 000、2 500 μmol/L;吉非替尼: 0、5、10、15、20、25 μmol/L),再将培养板放入CO2培养箱继续培养48 h。然后每孔加入MTT 试剂 20 μL于37℃培养2 h。翻板倒掉上清,加100 μL DMSO溶解。用分光光度计测定570 nm波长处光密度(D)值,计算细胞生存率。细胞生存率(%)=[(实验组D值-本底D值)/对照组D值]100%。 1.3 蛋白质印迹分析检测E-cadherin蛋白表达
提取蛋白并定量样本,将等量的蛋白样品(50 μg)加入聚丙烯酰胺凝胶,常规电泳、转膜。用含5%脱脂牛奶的PBS封闭NC膜,室温封闭60 min;加入一抗(1∶1 000)摇床上4℃过夜;PBS洗4次;加入二抗(1∶50 000)摇床上室温孵育60 min;PBS洗4次。最后加入ECL底物反应5 min,暗室显影,β-actin为内参。 1.4 转染实验
将已经获得的E-cadherin质粒转染耐药的肝癌细胞,转导2 d后将细胞转移至2个10 cm 培养皿,待细胞贴壁后换成含抗生素的培养液继续培养,直至阳性克隆肉眼可见。3周后开始挑阳性克隆,每个克隆的细胞分成2份,1份培养于6孔板,另1份培养在培养瓶中。收集培养于6孔板的细胞作蛋白质印迹分析以挑选E-cadherin上调幅度最大的阳性克隆,待筛选和鉴定完毕后仅保留最佳的克隆作后续MTT和蛋白质印迹分析实验。 1.5 统计学处理
应用SPSS13.0软件进行统计学分析。PD153035和吉非替尼两种EGFR-TKI药物的浓度与4种肝细胞癌细胞HepG2、BEL-7404、SK-HEP-1和MHCC97生存率的相关性采用直线相关分析法;E-cadherin表达阴性细胞SK-HEP-1转染E-cadherin目的基因后与转入空载体的肝癌细胞生存率的差异比较采用χ2检验。检验水准(α)为0.05。 2 结 果 2.1 MTT法检测细胞增殖结果
用MTT法测定不同浓度的EGFR-TKI PD153035(0、500、1 000、1 500、2 000、2 500 μmol/L)和吉非替尼( 0、5、10、15、20、25 μmol/L)对肝癌细胞HepG2、BEL-7404、MHCC97和SK-HEP-1生存率的影响,结果(图 1)显示,HepG2、BEL-7404细胞对PD153035和吉非替尼均较敏感,而MHCC97、SK-HEP-1细胞对这两种EGFR-TKI均表现为耐药。相关分析显示,PD153035 浓度和吉非替尼浓度与肝癌细胞HepG2、BEL7404的生存率之间均存在相关性(P<0.05),而这两种EGFR-TKI浓度与肝癌细胞MHCC97、SK-HEP-1的生存率之间无相关性(P>0.05)。

|
图 1 MTT法测定两种EGFR-TKI作用后各肝癌细胞的生存曲线
EGFR-TKI:表皮生长因子受体酪氨酸激酶抑制剂. A:PD153035; B:吉非替尼. n=6,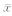 ±s ±s
|
蛋白质印迹分析结果(图 2)显示,HepG2、BEL-7404两种肝癌细胞中E-cadherin蛋白表达阳性;而SK-HEP-1和MHCC97两种肝癌细胞E-cadherin蛋白表达阴性。

|
图 2 蛋白质印迹分析检测各肝癌细胞中E-cadherin蛋白表达 |
蛋白质印迹分析结果(图 3)显示,转染E-cadherin目的基因的肝癌细胞SK-HEP-1中E-cadherin蛋白有表达,而转染空载体的肝癌细胞SK-HEP-1中无E-cadherin蛋白表达。

|
图 3 蛋白质印迹分析检测转染后肝癌细胞SK-HEP-1中E-cadherin蛋白表达 |
肝癌细胞SK-HEP-1转染E-cadherin目的基因后,MTT检测结果(图 4)显示,稳定表达E-cadherin能逆转SK-HEP-1细胞对PD153035和吉非替尼的敏感性。经χ2检验发现,SK-HEP-1细胞对PD153035、吉非替尼的敏感性与空载体组比较,差异均有统计学意义(P<0.05)。

|
图 4 MTT测定稳定表达E-cadherin后两种EGFR-TKI对肝癌细胞SK-HEP-1生存率曲线的影响
EGFR-TKI:表皮生长因子受体酪氨酸激酶抑制剂. A:PD153035; B:吉非替尼. *P<0.05与空载体组比较. n=6,±s
|
目前针对EGFR开展的分子靶向治疗有两大类[ 4,5 ]: 一类是进入细胞内的TKI,系与ATP竞争结合酪氨酸激酶并间接抑制其功能的小分子化合物。常见的EGFR-TKI包括吉非替尼、厄洛替尼(erlotinib)、PD153035。另一类是EGFR单克隆抗体,主要作用于EGFR的细胞外区域,与各种配体(如EGF、TGF-β)竞争结合并导致EGFR失去活性,包括西妥昔单抗(cetuximab) 和帕尼单抗 (panitumumab)。在临床应用方面TKI优于EGFR单克隆抗体,其疗效最早是在肺癌患者的治疗中得到肯定,尤其对传统化疗药物无效的部分患者仍然有效,以后逐渐应用于乳腺癌、胰腺癌、直肠癌以及盆腔和泌尿系统等肿瘤的治疗[ 6,7,8,9,10,11 ]。
本研究中,EGFR-TKI(PD153035和吉非替尼)对肝癌细胞HepG2、BEL-7404、SK-HEP-1和MHCC97抑制率的检测发现,肝癌细胞HepG2、BEL-7404中E-cadherin表达阳性并对EGFR-TKI治疗敏感,然而肝癌细胞SK-HEP-1和MHCC97中E-cadherin表达阴性,对EGFR-TKI治疗耐药。另外,在E-cadherin表达阴性细胞SK-HEP-1转染E-cadherin后发现EGFR-TKI治疗的敏感性上调。这说明E-cadherin在基因和蛋白水平的表达与EGFR-TKI治疗肝细胞癌耐药/敏感性存在相关性,并在其中可能扮演了重要作用,他们之间可能存在一些相互作用的机制。
E-cadherin为跨膜的单链多肽,由含氨基末端的膜外部分、跨膜区和含羧基末端的胞质内部分等组成。膜外部分负责细胞黏附,胞内部分与连环蛋白(α-catenin、β-catenin)形成复合物。EGFR和E-cadherin发生相互作用的空间基础是两者的亚细胞定位非常靠近,EGFR和E-cadherin共同存在于细胞间的黏附小带区。研究发现两者存在双向的作用关系[ 12 ]。在EGFR调节E-cadherin方面,E-cadherin的胞质内区与β-catenin的核心区连接成复合物,再连接到肌动蛋白的细胞骨架上,以维持细胞间的紧密连接。β-catenin是EGFR作用的底物,当EGFR配体(如EGF、TGF-β等)与EGFR结合后,具有酪氨酸激酶活性的EGFR胞质内区活化,引起E-cadherin与β-catenin复合物中的β-catenin磷酸化,导致β-catenin从复合体解离并使复合物从肌动蛋白上解离开来,从而降低细胞间的黏附性。因此,配体与EGFR的结合能促使E-cadherin表达的下调和EMT的发生[ 13 ]。在E-cadherin调节EGFR方面,E-cadherin是一个钙依赖的细胞膜表面黏附分子,当细胞环境从低钙向高钙环境变化时能激活E-cadherin的活化和EGFR的磷酸化[ 14,15 ]。此外,E-cadherin通过其细胞外的结构域和EGFR受体结合,还将减少后者的活动性以及与EGF结合的亲和力[ 16 ]。近年研究还发现,在E-cadherin突变的情况下,EGFR的磷酸化和内化(internalization)显著增强[ 17 ]。但是这些研究均未提及E-cadherin的正常功能、缺少或突变对EGFR靶向治疗敏感性的影响。在EGF等生长因子的刺激下,EGFR内化的速度加快,同时EGFR某些区域激活以后,加速蛋白降解,这些可能是机体内一种控制EGF信号强弱和持续性的机制。
另外,在E-cadherin调节EGFR的过程中,可能存在上游基因miRNA对E-cadherin的调控,不少研究发现miRNA对E-cadherin调控在EGFR分子靶向治疗中有重要作用。近期研究表明,EMT可能导致EGFR介导治疗的耐药。首先EMT改变是细胞在非编码RNA作为一个调节因子在细胞发展过程中起作用的一个发展过程,此过程伴有E-cadherin的丢失。Adam等[ 18 ]发现miRNAs在影响EMT方面的潜能,其介导的人膀胱肿瘤对EGFR治疗的耐药性有抑制效应,用miRNAs 矩阵列筛选及实时定量反转录聚合酶链反应来鉴定和验证9个膀胱肿瘤细胞系中涉及到EMT的miRNAs 的不同表达情况,发现miRNA-200家族中miRNAs的表达与上皮细胞形态及对EGFR阻断剂诱导膀胱肿瘤细胞的生长抑制的敏感性有很密切的联系。在稳定表达miRNA-200的间质细胞UMUC3中,E-cadherin表达水平明显上调,而ZEB1、ZEB2、ERRFI-1表达以及细胞转移能力下降,对EGFR阻断剂敏感性也提高。通过沉默或过表达ERRFI-1或miRNA-200而引起EGFR敏感性的变化也在UMUC5和T24细胞系中得到验证。认为miRNA-200家族成员可能参与了控制EMT的过程和对膀胱肿瘤EGFR靶向治疗的敏感性。同时认为miRNA-200的表达至少在一些间质性的膀胱癌细胞中可以恢复EGFR的依赖性,miRNA-200诱导的ERRFI-1靶向治疗可能是一个新的EGFR非依赖的调节剂。不少文献还报道miRNA-9[ 19 ]、miRNA-34a[ 20 ]等对E-cadherin及EMT有重要的调节作用,至于miRNA-9对 EGFR-TKI分子靶向治疗的调控方面,目前不清楚这些直接或者间接调节E-cadherin的miRNAs参与肿瘤调控和对EGFR-TKI靶向分子治疗的具体作用,需要进一步的研究才能确定瘤体中miRNA-9对E-cadherin和对其他靶点的作用机制。
以上这些可能是E-cadherin参与EGFR-TKI分子靶向治疗的一些调控机制,E-cadherin与EGFR-TKI双向作用的关系极其复杂,其具体作用的信号调控通路和如何逆转其耐药机制还有待进一步研究。
4 利益冲突
所有作者声明本文不涉及任何利益冲突。
| [1] | Saif M W.Colorectal cancer in review:the role of the EGFR pathway[J].Expert Opin Investig Drugs, 2010, 19:357-369. |
| [2] | Fratto M E, Santini D, Vincenzi B, Silvestris N, Azzariti A, Tommasi S, et al.Targeting EGFR in bilio-pancreatic and liver carcinoma[J].Front Biosci (Schol Ed), 2011, 3:16-22. |
| [3] | 王敬萍, 郑 华, 李宝兰, 付 瑜.EGFR基因突变与肺癌EGFR-TKI获得性耐药的研究进展[J].实用临床医药杂志, 2008, 12:12-15. |
| [4] | Okamoto I.Epidermal growth factor receptor in relation to tumor development:EGFR-targeted anticancer therapy[J].FEBS J, 2010, 277:309-315. |
| [5] | Lurje G, Lenz H J.EGFR signaling and drug discovery[J].Oncology, 2009, 77:400-410. |
| [6] | Thomson S, Buck E, Petti F, Griffin G, Brown E, Ramnarine N, et al.Epithelial to mesenchymal transition is a determinant of sensitivity of non-small-cell lung carcinoma cell lines and xenografts to epidermal growth factor receptor inhibition[J].Cancer Res, 2005, 65:9455-9462. |
| [7] | Yauch R L, Januario T, Eberhard D A, Cavet G, Zhu W, Fu L, et al.Epithelial versus mesenchymal phenotype determines in vitro sensitivity and predicts clinical activity of erlotinib in lung cancer patients[J].Clin Cancer Res, 2005, 11(24 Pt 1):8686-8698. |
| [8] | Black P C, Brown G A, Inamoto T, Shrader M, Arora A, Siefker-Radtke A O, et al.Sensitivity to epidermal growth factor receptor inhibitor requires E-Cadherin expression in urothelial carcinoma cells[J].Clin Cancer Res, 2008, 14:1478-1486. |
| [9] | Lee M Y, Chou C Y, Tang M J, Shen M R.Epithelial-mesenchymal transition in cervical cancer:correlation with tumor progression, epidermal growth factor receptor overexpression, and snail up-regulation[J].Clin Cancer Res, 2008, 14:4743-4750. |
| [10] | Buck E, Eyzaguirre A, Barr S, Thompson S, Sennello R, Young D, et al.Loss of homotypic cell adhesion by epithelial-mesenchymal transition or mutation limits sensitivity to epidermal growth factor receptor inhibition[J].Mol Cancer Ther, 2007, 6:532-541. |
| [11] | Fuchs B C, Fujii T, Dorfman J D, Goodwin J M, Zhu A X, Lanuti M, et al.Epithelial-to-mesenchymal transition and integrin-linked kinase mediate sensitivity to epidermal growth factor receptor inhibition in human hepatoma cells[J].Cancer Res, 2008, 68:2391-2399. |
| [12] | Andl C D, Rustgi A K.No one-way street:cross-talk between E-cadherin and receptor tyrosine kinase (RTK) signaling:a mechanism to regulate RTK activity[J].Cancer Biol Ther, 2005, 4:28-31. |
| [13] | Yasmeen A, Bismar T A, Al Moustafa A E.ErbB receptors and epithelial-cadherin-catenin complex in human carcinomas[J].Future Oncol, 2006, 2:765-781. |
| [14] | Pece S, Gutkind J S.Signaling from E-cadherins to the MAPK pathway by the recruitment and activation of epidermal growth factor receptors upon cell-cell contact formation[J].J Biol Chem, 2000, 275:41227-41233. |
| [15] | Heijink I H, Kies P M, Kauffman H F, Postma D S, van Oosterhout A J, Vellenga E.Down-regulation of E-cadherin in human bronchial epithelial cells leads to epidermal growth factor receptor-dependent Th2 cell-promoting activity[J].J Immunol, 2007, 178:7678-7685. |
| [16] | Qian X, Karpova T, Sheppard A M, McNally J, Lowy D R.E-cadherin-mediated adhesion inhibits ligand-dependent activation of diverse receptor tyrosine kinases[J].EMBO J, 2004, 23:1739-1748. |
| [17] | Bremm A, Walch A, Fuchs M, Mages J, Duyster J, Keller G, et al.Enhanced activation of epidermal growth factor receptor caused by tumor-derived E-cadherin mutations[J].Cancer Res, 2008, 68:707-714. |
| [18] | Adam L, Zhong M, Choi W, Qi W, Nicoloso M, Arora A, et al.MiR-200 expression regulates epithelial-to-mesenchymal transition in bladder cancer cells and reverses resistance to epidermal growth factor receptor therapy[J].Clin Cancer Res 2009, 15:5060-5072. |
| [19] | Liu S, Kumar S M, Lu H, Liu A, Yang R, Pushparajan A, et al.MicroRNA-9 up-regulates E-cadherin through inhibition of NF-κB1-Snail1 pathway in melanoma[J].J Pathol, 2012, 226:61-72. |
| [20] | Nalls D, Tang S N, Rodova M, Srivastava R K, Shankar S.Targeting epigenetic regulation of miR-34a for treatment of pancreatic cancer by inhibition of pancreatic cancer stem cells[J].PLoS One, 2011, 6:e24099. |