 2019, Vol. 36
2019, Vol. 36 

2. General Hospital of Wanbei Coal-electric Group, Suzhou 234011, China
2. 皖北煤电集团总医院, 安徽 宿州 234011
In eukaryotes, the post-translational modifications (PTMs) of histones play an essential role in the regulation of chromatin structural dynamics and gene expression. Histone methylation, one of the key epigenetic modifications, usually occurs on the lysine (K) or arginine (R) residues at the N-terminal flexible tails of histones, and regulates transcriptional activation or inactivation of specific genes[1, 2]. The nuclear receptor binding SET domain (NSD) family has three SET domain-containing methyltransferases, i.e., NSD1, NSD2, NSD3[3]. NSD proteins specifically methylate K36 of histone H3 on lysine 36 (H3K36). Importantly, such PTM is associated with multiple human diseases including cancers[4-7]. Each NSD protein contains a SET domain, a PWWP domain and PHD fingers[8]. The catalytic SET domain is an evolutionarily conserved motif with 130~150 amino acids, while the PWWP and PHD domains are readers of other proteins or nucleic acids.
NSD1 mutations are closely relative to multiple human diseases. For instance, NSD1 mutations cause Sotos syndrome, which has the symptom of childhood overgrowth, learning disability, and mental retardation[4-6, 9, 10]. NSD1-NUP98 fusion protein, resulting from chromosomal translocations, is associated with childhood acute myeloid leukemia[7, 11]. The inactivating mutation of NSD1 is linked to multiple cancers as well[9, 12]. Therefore, it is of significance to discover small molecule inhibitors to probe the biologic functions of NSD1 and to exploit the potential therapeutic treatment. The SET domains are drug gable targets as have been validated by the inhibitors of DOT1L/KMT4, EZH2/KMT6, and G9a[13-15]. However, to our best knowledge, no small molecule inhibitors of the NSD1 has yet been identified.
Fragment-based drug discovery (FBDD) has been proven effective and fruitful for the past 20 years, even for challenging protein-protein interaction (PPI) targets with shallow pockets[16]. FBDD starts from low-molecular-weight compounds (fragments), and usually directly detects the weak interactions between targets and fragments by using biophysical methods, e.g., surface plasmon resonance (SPR), fluorescence spectroscopy, nuclear magnetic resonance (NMR), and X-ray crystallography[17, 18]. The combination of protein-observed and ligand-observed NMR spectroscopy enabled robust detection of ambiguous weak interactions, due to NMR nature of multi-valued structural output[19, 20].
Here, we applied an automated NMR fragment-based screening (FBS) and identified three weak binders of the NSD1 SET domain. To enhance the screening throughput, we first identified feasible hits from the fragment cocktails by using ligand-observed spectra, e.g., saturation transfer difference (STD) and WaterLOGSY. These experiments were then reproduced for each individual hit. The three specific binders were further validated by the dose-dependent chemical shift perturbations (CSPs) of 15N-labeled NSD1 SET domain. We finally analyzed the binding mode of the hit using molecular docking.
1 Materials and methods 1.1 Cloning, protein expression and purificationDNA encoding NSD1 SET domain (residues 1 852~2 082) was cloned into the pGEX4T-1 vector (GE Healthcare, Shanghai, China), which was modified with tobacco etch virus (TEV) cleavage sites. The construct was then transformed into Escherichia coli BL21(DE3)-RIL. Cells were grown in minimal medium supplemented with 15NH4Cl for NMR samples. The protein expression was generally induced at OD600 of 0.8~1.2 using 0.5 mmol/L isopropyl β-D-thiogalactos (IPTG) at 16 ℃ for 24 h. The glutathione S-transferase (GST)-tagged proteins were purified from pretreated bacterial lysates using GSTrap FF (GE Healthcare, Shanghai, China), then treated with TEV to cleave the N-terminal GST tag. Protein was purified further by size exclusion chromatography using a HiLoad 16/60 Superdex 200 column (GE Healthcare, Shanghai, China). The purified protein was stored in Tris buffer [20 mmol/L Tris, 200 mmol/L NaCl, 2 mmol/L tris(2-carboxyethyl)phosphine (TCEP) at pH 6.8].
1.2 NMR fragment-based screeningThe whole process of NMR fragment-based screening was performed at 25 ℃ on a 700 MHz spectrometer (Agilent Technologies Santa Clara, CA, USA) equipped with a 96-well autosampler and a cryoprobe[19]. In brief, the screening samples were prepared containing 0.4 mmol/L compounds in DMSO-d6 solution (cocktails for primary screening), 50% D2O and 10 μmol/L unlabeled protein. The binding hits were identified from the primary screening in cocktail and secondary screening for each individual compound based on the signals of STD and WaterLOGSY spectra.
The STD experiment was acquired using the acquisition time of 1 s, 32 dummy scans, and relaxation delay of 0.1 s, followed by a 2 s Gauss pulse train with the irradiation frequency at 20.7 ppm or 250 ppm alternatively. The total acquisition time was 15 min with 256 scans. WaterLOGSY was acquired for 15.1 min with 1 s acquisition time, 1 s relaxation and 1.3 s NOE mixing time and 256 scans.
1.3 NMR CSP calculationThe 15N-labeled NSD1 proteins were concentrated to 0.1 mmol/L in Tris buffer. The compounds were then titrated into 15N-labeled NSD1. The HSQC spectra were collected for each titration points on Agilent 700 MHz spectrometer at the ligand/protein molar ratios of 0, 0.25, 0.5, 1, 2, 5, respectively. The binding constant was best-fitted using the following equation assuming a 1:1 binding mode[24]
| $ \begin{array}{l} \mathit{\Delta }\delta = \mathit{\Delta }{\delta _{\max }}\\ \left\{ {{K_D} + {{[P]}_t} + {{[L]}_t} - {{\left[ {{{\left( {{K_D} + {{[P]}_t} + {{[L]}_t}} \right)}^2} - \left( {4*{{[P]}_t}{{[L]}_t}} \right)} \right]}^{1/2}}} \right\}/\left( {2{{[P]}_t}} \right) \end{array} $ | (1) |
where [P]t and [L]t are total concentrations of ligand and protein, respectively, Δδ is the chemical shift change with respect to the free state; maximum chemical shift change (Δδmax) and binding affinity (KD) were best-fits from the dose-dependent chemical shift changes.
2 Results and discussionNSD1 is a 2 969 amino-acid protein containing four PHD fingers, two PWWP domains, and one catalytic SET domain [Fig. 1(a)]. Although the crystal structure of NSD1 containing AWS, SET and post-SET sequence motifs (amino acids 1 852~2 082) has been resolved (PDB code: 3OOI), little is known about its solution structure and dynamics. The 1H-15N heteronuclear single quantum coherence (HSQC) spectrum is powerful to assess the protein folding and aggregation status. We hence expressed and purified the 15N-labeled protein fused to a GST tag with TEV protease cleavage site [Fig. 1(b) and 1(c)]. The transverse relaxation optimized HSQC (TROSY-HSQC) spectrum suggests that the NSD1 SET domain is mono-dispersed [Fig. 1(d)], suitable for following FBS.

|
Fig. 1 (a) The domain structure of NSD1 containing 2 969 amino acids. The black box represents each domain with the domain name annotated; (b) SDS-PAGE of the NSD1 SET domain. Lanes 1~6 represent marker, elution of NSD1, washing, flow through, supernatant, and precipitation, respectively; (c) The UV spectrum at wave length of 280 nm of NSD1 SET (domain 1 852~2 082 amino acid) purified by 16/60 Superdex 200 size exclusion columns; (d) The 1H-15N TROSY-HSQC spectrum of NSD1 SET domain |
Our automated NMR FBS[19] was utilized to uncover hits of the NSD1 SET domain. NMR FBS integrates the ligand-observed and protein-observed experiments[17]. The protein-observed NMR FBS method provides valuable information of binding sites and affinity. However, protein-observed NMR FBS requires a significant amount of 15N-labeled proteins and a high quality of the 2D 1H-15N HSQC spectrum. Conversely, the ligand-observed NMR FBS experiments record the changes of ligand signals in the presence of a small portion of the unlabeled protein. STD and WaterLOGSY are the most widely used spectra in NMR FBS. To enhance the throughput of primary screening, we pooled the fragment library of 890 compounds into 89 cocktails with 10 compounds each. The compound and protein concentrations were set to 0.4 and 0.01 mmol/L, respectively. A total of 16 hits were identified as they presented signals in STD and inverted sign of signals in WaterLOGSY [Fig. 2(a)]. The 16 hits were further filtered by a secondary screening using the same set of aforementioned experiments for each individual compound [Fig. 2(b)]. Such a cross validation procedure produced 7 hits.

|
Fig. 2 Ligand-observed NMR fragment-based screening against the NSD1 SET domain. (a) The primary screening over a typical cocktail. The top 10 spectra are the reference 1H NMR spectra of each individual component of the cocktail, and the bottom 3 are WaterLOGSY, STD and the 1H NMR spectra of the cocktail; (b) The secondary screening for hit 1 alone, using the same set of screening spectra |
The hits identified from the above ligand-observed FBS were then validated by CSPs of the 15N labeled NSD1 SET domain. The hits were titrated to 0.12 mmol/L 15N-labeled NSD1 SET domain at a final ligand/protein molar ratio varying from 0 to 5 [Fig. 3(a)]. Three hits perturbed a common set of residues, indicating that these hits bind specifically to the NSD1 SET domain. The binding affinities determined from the dose-dependent CSPs [Fig. 3(b)] were 0.12, 0.54 and 1.22 mmol/L for the three hits, respectively (Table 1). The hit rate and the reasonable ligand efficiencies (binding energy per non-proton atom) indicate the NSD1 SET domain is a drug gable target.

|
Fig. 3 Chemical shift perturbations induced by the fragment screening hit 1. (a) Chemical shift perturbations of 15N-labeled NSD1 SET domain upon titration of hit 1 at the indicated ligand/protein molar ratios; (b) The affinity of hit 1 to NSD1 SET domain is best-fitted from the dose-dependent chemical shift changes (Δδ). Δδ was calculated as [(ΔδH)2+(ΔδN/5)2]1/2, where ΔδH and ΔδN denote the chemical shift changes of 1H and 15N, respectively |
| Table 1 Structure and affinities of fragment screening hits of NSD1 SET domain |
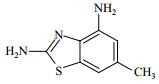
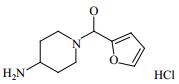
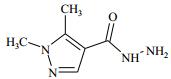
The target-ligand interaction modes are essential for rational drug discovery. However, we failed to crystallize the NSD1 SET domain complexes, probably due to the low binding affinities of the hits. We hence simulated the complex structural models using the molecular docking software AutoDock, in a way as has been published previously[21-23]. Notably, the lowest-energy docking pose demonstrated compound 1 bound to the AdoMet binding pocket [Fig. 4(a) and 4(b)] and formed hydrogen bonds with residues H2021 and K2071, respectively [Fig. 4(c)]. This interaction pattern is consistent with that of AdoMet as revealed in the complex crystal structure[8] [Fig. 4(d)]. We recently demonstrated that paramagnetic NMR spectroscopy facilitated the filtration of the best-fit docking pose[21-23]. We are now working on the backbone chemical shift assignment of NSD1 SET domain and subsequent acquisition of NMR structural restraints.

|
Fig. 4 Docking modes of hit 1 in comparison with the natural substrate AdoMet of the NSD1 SET domain. (a) Electrostatic surface representation of NSD1 SET domain (PDB code: 3OOI) with hit 1 (carbon atoms in yellow sticks), superimposed with the crystal structure in complex with AdoMet (carbon atoms in green sticks); (b) Binding site topology of the NSD1 SET domain in complex with AdoMet (green) and hit 1 (yellow), respectively; (c) The lowest-energy docking pose suggests the interaction details of hit 1, which may form hydrogen bonds (red dotted lines) with H2021 and K2071, respectively; (d) The binding pattern revealed from the crystal structure of NSD1 in complex with AdoMet |
NSD1 is a potential therapeutic target for multiple human cancers and Sotos syndrome. We discovered new hits by using NMR FBS against the NSD1 SET domain. Three specific binders were confirmed by NMR CSPs. The molecular docking poses exhibited a similar pattern to the natural substrate AdoMet. Our study shall enable the following structure-guided hit-to-lead evolution targeting the NSD1 SET domain.
| [1] | BAYLIN S B, JONES P A. A decade of exploring the cancer epigenome-biological and translational implications[J]. Nat Rev Cancer, 2011, 11(10): 726-734. DOI: 10.1038/nrc3130. |
| [2] | COLE P A. Chemical probes for histone-modifying enzymes[J]. Nat Chem Biol, 2008, 4(10): 590-597. DOI: 10.1038/nchembio.111. |
| [3] | HAN J Y, LEE I G, JANG W, et al. Identification of a novel de novo nonsense mutation of the NSD1 gene in monozygotic twins discordant for Sotos syndrome[J]. Clin Chim Acta, 2017, 470: 31-35. DOI: 10.1016/j.cca.2017.04.025. |
| [4] | BERDASCO M, ROPERO S, SETIEN F, et al. Epigenetic inactivation of the Sotos overgrowth syndrome gene histone methyltransferase NSD1 in human neuroblastoma and glioma[J]. Proc Natl Acad Sci U S A, 2009, 106(51): 21830-21835. DOI: 10.1073/pnas.0906831106. |
| [5] | LU T, JACKSON M W, WANG B L, et al. Regulation of NF-kappaB by NSD1/FBXL11-dependent reversible lysine methylation of p65[J]. Proc Natl Acad Sci U S A, , 107(1): 46-51. DOI: 10.1073/pnas.0912493107. |
| [6] | PERI S, IZUMCHENKO E, SCHUBERT A D, et al. NSD1- and NSD2-damaging mutations define a subset of laryngeal tumors with favorable prognosis[J]. Nat Commun, 2017, 8(1): 1772. DOI: 10.1038/s41467-017-01877-7. |
| [7] | SHIBA N, ICHIKAWA H, TAKI T, et al. NUP98-NSD1 gene fusion and its related gene expression signature are strongly associated with a poor prognosis in pediatric acute myeloid leukemia[J]. Genes Chromosomes Cancer, 2013, 52(7): 683-693. |
| [8] | QIAO Q, LI Y, CHEN Z, et al. The structure of NSD1 reveals an autoregulatory mechanism underlying histone H3K36 methylation[J]. J Biol Chem, 2011, 286(10): 8361-8368. DOI: 10.1074/jbc.M110.204115. |
| [9] | SU X P, ZHANG J P, MOUAWAD R, et al. NSD1 inactivation and SETD2 mutation drive a convergence toward loss of function of H3K36 writers in clear cell renal cell carcinomas[J]. Cancer Res, 2017, 77(18): 4835-4845. DOI: 10.1158/0008-5472.CAN-17-0143. |
| [10] | VISSER R, LANDMAN E B, GOEMAN J, et al. Sotos syndrome is associated with deregulation of the MAPK/ERK-signaling pathway[J]. PLoS One, 2012, 7(11): e49229. DOI: 10.1371/journal.pone.0049229. |
| [11] | WANG G G, CAI L, PASILLAS M P, et al. NUP98-NSD1 links H3K36 methylation to Hox-A gene activation and leukaemogenesis[J]. Nat Cell Biol, 2007, 9(7): 804-812. DOI: 10.1038/ncb1608. |
| [12] | TATTON-BROWN K, RAHMAN N. The NSD1 and EZH2 overgrowth genes, similarities and differences[J]. Am J Med Genet C Semin Med Genet, 2013, 163C(2): 86-91. |
| [13] | CHANG Y Q, ZHANG X, HORTON J R, et al. Structural basis for G9a-like protein lysine methyltransferase inhibition by BIX-01294[J]. Nat Struct Mol Biol, 2009, 16(3): 312-317. DOI: 10.1038/nsmb.1560. |
| [14] | YAO Y, CHEN P H, DIAO J S, et al. Selective inhibitors of histone methyltransferase DOT1L: design, synthesis, and crystallographic studies[J]. J Am Chem Soc, 2011, 133(42): 16746-16749. DOI: 10.1021/ja206312b. |
| [15] | MCCABE M T, OTT H M, GANJI G, et al. EZH2 inhibition as a therapeutic strategy for lymphoma with EZH2-activating mutations[J]. Nature, 2012, 492(7427): 108-112. DOI: 10.1038/nature11606. |
| [16] | ZHANG X Q, SONG F B, KUO G H, et al. Optimization of a pyrazole hit from FBDD into a novel series of indazoles as ketohexokinase inhibitors[J]. Bioorg Med Chem Lett, 2011, 21(16): 4762-4767. DOI: 10.1016/j.bmcl.2011.06.067. |
| [17] | MA R S, WANG P C, WU J H, et al. Process of fragment-based lead discovery-A perspective from NMR[J]. Molecules, 2016, 21(7): E854. DOI: 10.3390/molecules21070854. |
| [18] | WHITTAKER M. Picking up the pieces with FBDD or FADD: invest early for future success[J]. Drug Discov Today, 2009, 14(13/14): 623-624. |
| [19] | GAO J, MA R S, WANG W, et al. Automated NMR fragment based screening identified a novel interface blocker to the LARG/RhoA complex[J]. PLoS One, 2014, 9(2): e88098. DOI: 10.1371/journal.pone.0088098. |
| [20] | LIU J Y, ZHANG S Y, LIU M Q, et al. Structural plasticity of the TDRD3 Tudor domain probed by a fragment screening hit[J]. FEBS J, 2018, 285(11): 2091-2103. DOI: 10.1111/febs.2018.285.issue-11. |
| [21] | GAO J, LIANG E, MA R S, et al. Fluorine pseudocontact shifts used for characterizing the protein-ligand interaction mode in the limit of NMR intermediate exchange[J]. Angew Chem Int Ed Engl, 2017, 56(42): 12982-12986. DOI: 10.1002/anie.201707114. |
| [22] | XU D F, LI B, GAO J, et al. Ligand proton pseudocontact shifts determined from paramagnetic relaxation dispersion in the limit of NMR intermediate exchange[J]. J Phys Chem Lett, 2018, 9(12): 3361-3367. DOI: 10.1021/acs.jpclett.8b01443. |
| [23] | LIU J Y, GAO J, LI F D, et al. NMR characterization of weak interactions between RhoGDI2 and fragment screening hits[J]. Biochim Biophys Acta Gen Subj, 2017, 1861(1 Pt A): 3061-3070. |
| [24] | WILLIAMSON M P. Using chemical shift perturbation to characterise ligand binding[J]. Prog Nucl Magn Reson Spectrosc, 2013, 73: 1-16. DOI: 10.1016/j.pnmrs.2013.02.001. |