 2017, Vol. 34
2017, Vol. 34 

2. 上海纽迈电子科技有限公司, 上海 200062;
3. 双钱集团上海轮胎研究所有限公司, 上海 200237;
4. 同济大学 材料科学与工程学院, 上海 201804
2. Shanghai Niumag Electronic Technology Co., Ltd, Shanghai 200062, China;
3. Double Coin Group Shanghai Tire Research Institute Co., Ltd, Shanghai 200237, China;
4. School of Materials Science and Engineering, Tongji University, Shanghai 201804, China
交联密度是指交联聚合物里面交联键的多少,一般用交联点之间的相对分子质量(Mc)来表示[1].交联密度对于橡胶的弹性、拉伸强度、断裂伸长率、硬度有很大的影响[2].很多测试技术可用于橡胶交联密度的测量,其中力学测试法[3]是根据橡胶的弹性理论和唯象理论来诠释硫化胶的交联密度和测试结果之间的关系,并发展了几种理想的分子模型[4-6].但这些模型的理论和实验结果之间会由于链缠结、网络缺陷及网络的不均匀性等因素而存在一些差异,因此力学法并不能提供关于硫化胶网络结构的全部可靠信息[7, 8],而且这种传统的方法存在测试周期长、影响因素复杂、误差大等缺点[1].
1956年,Mullins[9]报道了基于Flory-Rehner(FR)理论的平衡溶胀法测量硫化天然橡胶的交联密度.交联点的存在使得橡胶无法溶解到溶液中,只能溶胀.溶胀平衡时的体积与交联密度有关,可以通过平衡前后体积的变化来计算橡胶的交联密度.1971年,Gennes[10]报道了基于核磁共振(NMR)技术的橡胶交联密度测量方法.交联点间分子量的大小会对链段的运动产生影响,因此通过分析高分子链的运动可以研究硫化胶的交联密度.Kuhn等[11, 12]在Gennes的基础理论和实验结果的基础上对硫化胶的网络结构和交联密度进行了详细的研究,结果证明NMR横向弛豫参数和传统的表征硫化胶交联密度的方法之间有良好的相关性,并建立了NMR弛豫参数和硫化胶结构之间的关系.该方法可以克服传统方法的部分缺点[13].在众多基于NMR的橡胶交联密度测量方法中,1H双量子(DQ)NMR[14-16]以及基于1H Hahn回波的“1H CPMG”(Carr-Purcell-Meiboom-Gill)回波法[17, 18]是被应用最广泛的两种方法.Kuhn等[11, 12]提出,通过测量1H横向弛豫可以获得橡胶的交联密度. 1986年,Baum和Pines[17]开发了1H DQ脉冲序列,通过测量双量子相干效率可以获得所测体系中残余偶极相互作用Dres.随后的工作表明通过测量橡胶体系的Dres可以获得橡胶的交联密度[24, 25].
本文将系统地比较1H DQ NMR、1H Hahn回波和1H CPMG回波三种NMR方法在测量橡胶交联密度方面的差异,并讨论这些差异的来源.在此基础上,本文还将进一步研究1H CPMG回波方法中不同回波时间对测量值的影响,探讨如何改善1H CPMG回波方法以实现对橡胶交联密度更为可靠地测量.
1 实验部分 1.1 仪器与试剂仪器:AVANCE Ⅲ 300型NMR谱仪(Bruker)、VTMR20-010V-T型交联密度测试仪(上海纽迈电子科技有限公司)、XK-250型开炼机(上海橡胶机械一厂)、XLB-D型平板硫化机(湖州顺力橡胶机械公司).
药品与试剂:泰国20#标准胶(双钱集团上海轮胎研究所有限公司)、硬脂酸(江苏昆宝集团有限公司)、氧化锌(京华氧化锌有限公司)、炭黑(上海卡博特有限公司)、硫磺(江苏宏泰橡胶助剂有限公司),甲苯(昆山申瑞化工有限公司).
1.2 实验过程 1.2.1 试样准备本文所用的橡胶为泰国标准胶.先将橡胶在XK-250型开炼机上塑炼,待胶料表面光滑后,依次加入硬脂酸、氧化锌、炭黑和其他配合剂,最后加入硫磺,打三角包,薄通后出片,硫磺份数为7 phr(每100 g橡胶中加入7 g的硫磺).停放4 h后,用XLB-D型平板硫化机进行硫化,硫化温度为150 ℃,分别在硫化3 min、7 min、10 min、30 min以及60 min时取出部分胶料以备后续测试.
1.2.2 1H横向弛豫测量橡胶交联密度图 1为高分子网络模型示意图[20, 21],(a)为两端都受到交联点束缚的交联链,运动性很差;(b)为一端受到束缚的悬垂链,运动性好于交联链;(c)为体系中未受到束缚的自由链,运动性最好.对于橡胶材料来说,交联分为化学交联和物理交联,化学交联是链与链之间通过化学键连接,如硫化胶中的碳-硫键,物理交联则是由链缠结所产生的交联点.

|
图 1 高分子网络模型:(a)交联链;(b)悬垂链;(c)自由链(溶剂小分子);(x)化学交联;(y)物理交联(缠结) Figure 1 The model of polymer network: (a) Inter-crosslinked chain; (b) Dangling chain end; (c) Free chain (sol part); (x) Chemical crosslink; (y) Physical loop (entanglement) |
图 2以硫化3 min橡胶的静态1H NMR谱为例,展示了橡胶样品的静态1H NMR谱线的特征.该谱图在Bruker AVANCE Ⅲ 300型NMR谱仪上利用90°单脉冲实验获得.实验条件:90°脉宽为4 μs,等待时间为2 s,累加次数为4,实验温度为90 ℃.图 2中的信号是由一个较强的主峰与主峰左侧的一个较弱信号组成.这些信号来源于橡胶分子的-CH3、-CH2-和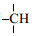

|
图 2 硫化3 min橡胶的静态1H NMR谱图 Figure 2 Static 1H NMR spectrum of a rubber with curing time of 3 min |
通常,橡胶在玻璃化转变温度(Tg)以上时,质子横向弛豫可以通过基于单条分子链模型和管状模型的理论方程(1)式来表示[21]:
| $ \frac{{{M}_{t}}}{{{M}_{0}}}=A\exp (-t/{{T}_{2}}-0.5q{{M}_{2}}{{t}^{2}})+B\exp (-t/{{T}_{2}})+C\exp (-t/{{T}_{2\rm{s}}})+{{A}_{0}} $ | (1) |
(1) 式中:
由于溶胶分子的含量较少,为了简化拟合的过程,可以假设溶胶含量为0,简化后为(2)式:
| $ \frac{{{M}_{t}}}{{{M}_{0}}}=A\exp (-t/{{T}_{2}}-0.5q{{M}_{2}}{{t}^{2}})+B\exp (-t/{{T}_{2}})+{{A}_{0}} $ | (2) |
根据Gotlib[23, 24]的观点,在玻璃化转变温度之上的二阶矩的比率因子q,可用于估计交联分子链的平均分子量Mc.Mc的计算见(3)式:
| $ {{M}_{\rm{c}}}=\frac{3c{{M}_{\rm{ru}}}}{5n\sqrt{q}} $ | (3) |
(3) 式中:c= 7.2,为Kuhn链段内的主链键数;Mru是重复单元内的摩尔质量,单位为g/mol;n为重复单元内的主链键数,Mru/n = 68/4 (g/mol).
对于单位体积橡胶基质中弹性活性网络链的数量(VNMR),可由(4)式计算得到:
| $ {{V}_{\rm{NMR}}}=A\rho /{{M}_{\rm{c}}} $ | (4) |
(4) 式中:A为(1)式中交联链的含量;ρ为天然橡胶的密度.
利用NMR谱仪测量橡胶的横向弛豫有两种方法,分别为1H Hahn回波法和1H CPMG回波法.本文在Bruker AVANCE Ⅲ 300型NMR谱仪上利用1H Hahn回波序列测量橡胶的质子横向弛豫,使用4 mm静态探头,90°和180°脉宽分别为4 μs和8 μs,等待时间为2 s,累加次数为8,实验温度为90 ℃.在VTMR20-010V-T型低场NMR交联密度测试仪上利用1H CPMG回波序列测量橡胶的质子横向弛豫,磁场强度为0.517 T,90°和180°脉宽分别为2.8 μs和5.8 μs,等待时间为2 s,累加次数为8,实验温度为90 ℃.
1.2.3 1H DQ NMR测量橡胶交联密度常规的1H DQ NMR实验的脉冲序列是由Baum和Pines于1986年发表[18].整个脉冲序列由两部分组成,分别为激发组合脉冲和恢复组合脉冲,这两部分的脉冲组合是相同的,只是相位有所不同.通过第一部分脉冲能激发天然橡胶体系内所有的偶量子数相干,而第二部分脉冲则将所有高阶相干恢复到可观测状态.之后通过相位循环得到两组数据,分别是参考信号和双量子信号,用IDQ和Iref来表示.IDQ包括受偶极相互作用影响的信号和所有(4n+2)阶相干的信号,而Iref则是由未耦合组分(各向同性运动部分)和4n阶相干信号组成.这样(IDQ+Iref)就包含了受弛豫作用影响的所有信号,但是必须要减去信号中由运动性较好的末端链所带来的影响,文献[23]详细地说明了数据处理方法.归一化的DQ累积数据可用(5)式表示:
| $ {{I}_{n\rm{DQ}}}=\frac{{{I}_{\rm{DQ}}}}{{{I}_{\rm{DQ}}}+{{I}_{\rm{ref}}}-f\exp (-2{{\tau }_{\rm{DQ}}}/{{T}_{2, \ \rm{tail}}})} $ | (5) |
其中
根据静态二阶矩近似理论,当InDQ≤0.45时,归一化的DQ累积曲线可用(6)式进行描述[24-26]:
| $ {{I}_{n\rm{DQ}}}({{D}_{\rm{res}}})=\frac{1}{2}\left[1-\exp (-\frac{2}{5}{{D}_{\rm{res}}}^{2}{{\tau }_{\rm{DQ}}}^{2}) \right] $ | (6) |
其中Dres是表观偶极相互作用参数,代表的是许多不同自旋的偶极相互作用的平均值,与交联点间的分子量Mc成反比.对天然橡胶来说,Dres和Mc的关系如(7)式[27, 28]:
| $ {{M}_{\rm{c}}}=\frac{617\ \rm{Hz}}{{{D}_{\rm{res}}}/2\pi }\rm{ kg/mol} $ | (7) |
在本文的工作中,1H DQ NMR实验在Bruker AVANCE Ⅲ 300型NMR谱仪进行,实验温度为90 ℃,磁体场强为7.04 T,使用4 mm静态探头,90°和180°脉宽分别为4 μs和8 μs,等待时间为2 s,累加次数为16,双量子时间间隔为400 μs.
1.2.4 平衡溶胀法测量橡胶交联密度交联聚合物在溶剂中不能溶解,但是能发生一定程度的溶胀,溶胀程度取决于聚合物的交联度,FR理论[29]可以用来分析橡胶的平衡溶胀实验.Cohen等[30]给出了计算交联密度的(8)式:
| $ {{M}_{\rm{c}}}=-\frac{{{\rho }_{\rm{p}}}{{V}_{\rm{s}}}{{\phi }_{\rm{p}}}^{1/3}}{\ln (1-{{\phi }_{\rm{p}}})+{{\phi }_{\rm{p}}}+\chi {{\phi }_{\rm{p}}}^{2}} $ | (8) |
(8) 式中:ρp为聚合物的密度,Vs为溶剂的摩尔体积,ϕ=Vp/V为溶胀平衡时聚合物的体积分数,χ为Flory-Huggins相互作用常数,表征聚合物和溶剂相互作用大小.
以下为本文中通过平衡溶胀法测交联密度的标准流程[9]:将厚约2 mm、重约0.2 g的橡胶样品放入装有15 mL甲苯的磨口试管中,盖紧瓶盖,放在25 ℃的恒温水槽中溶胀,每隔一定时间将样品取出,用滤纸迅速吸干表面的溶剂,立即放入去皮的置于天平中的磨口烧瓶中称量,直到最后两次的质量差在5 mg之内,则认为橡胶溶胀平衡,通过溶胀度可计算橡胶的交联密度.
2 结果与讨论 2.1 通过1H DQ NMR测量橡胶交联密度1H DQ NMR实验所测样品是硫化时间为3 min、7 min、10 min、30 min和60 min的橡胶.图 3(a)为硫化时间为3 min胶料的1H DQ NMR谱图.其中,圆点为测得的双量子信号IDQ,方点为参考信号Iref.图 3(b)将Iref与IDQ之差与双量子演化时间作图,由图中我们可以看到不同的弛豫行为,前半段信号弛豫较快,且信号以非指数形式弛豫,为交联链段的弛豫信号.后半段弛豫较慢,且信号以指数形式弛豫,为未交联链段的弛豫信号.由于未交联组分的非弹性缺陷信号会使得交联密度的测量产生误差,因此我们需要扣除这部分的信号.在数据处理过程中,我们选取了后半段数据进行拟合,拟合方程为(9)式,式中y由Iref与IDQ作差所得,拟合得到的非弹性组分含量f及质子横向弛豫时间(T2, tail)见表 1.
| $ y=f\exp (-2{{\tau }_{\rm{DQ}}}/{{T}_{2, \ \rm{tail}}}) $ | (9) |

|
图 3 一个橡胶样品的1H DQ NMR实验:(a)双量子信号(IDQ)和参考信号(Iref)曲线;(b) Iref与IDQ的差异曲线(Iref-IDQ).该样品的硫化时间为3 min Figure 3 1H DQ NMR of a rubber sample: (a) The DQ build-up (IDQ) and the reference (Iref) curves; (b) The difference (Iref-IDQ) curve. The sample has a curing time of 3 min |
| 表 1 1H DQ NMR的拟合参数及计算的Mc Table 1 The parameters used in the 1H DQ NMR data fitting and the yielded Mc |
将拟合得到的f及T2, tail代入(5)式,可得到归一化后的DQ累积数据InDQ随双量子演化时间的变化(图 4).由图可知,归一化的双量子信号随着双量子演化时间的变大而逐渐变大,最后到达一个平台.对图 4的数据可用(6)式进行拟合得到残余偶极相互作用Dres,通过(7)式可计算交联点间的分子量Mc.通过上述方法我们对硫化时间不同的样品进行了测量,这些样品的Dres和Mc见表 1.表中数据的误差值由数据拟合过程给出.

|
图 4 归一化的DQ曲线 Figure 4 The normalized DQ curve |
分析表 1中的数据我们发现,Mc随着硫化时间的增加而逐渐变小,说明此时交联点不断生成,交联密度变大.当硫化时间达到10 min时,Mc达到最小,之后Mc变大.表明此时硫磺消耗完全,硫化反应停止,出现了过硫反应.随着过硫反应的进行,材料中的部分交联键开始断裂,交联密度逐渐变小.在拟合中我们发现,对于所有样品而言,非弹性缺陷含量几乎一样,在10%左右,但是随着硫化时间的增加,非弹性缺陷组分的质子横向弛豫时间(T2, tail)整体在变小.我们推测产生这一现象的原因是交联密度的增加一定程度上限制了小分子的运动.
2.2 通过1H Hahn回波测量橡胶交联密度1H Hahn回波可以测量质子横向弛豫,因此常被用于橡胶交联密度的测定.我们利用1H Hahn回波序列对硫化时间为3 min、7 min、10 min、30 min和60 min的橡胶样品的质子横向弛豫进行了测量,在图 5中,我们以硫化3 min的胶料为例展示了1H Hahn回波测量的质子横向弛豫,以及如何通过(1)式对曲线进行拟合获得交联密度.1H Hahn回波法本质上也是测量体系的残余偶极相互作用,不同于1H DQ NMR,1H Hahn回波最终获得的是二阶矩的比率因子q.通过将q带入(3)式,即可计算网链间分子量Mc.我们使用上述方法对硫化时间不同的胶料进行了测量,表 2列出了交联链的比例A、悬尾链的比例B、溶胶的比例C、交联链和悬尾链的质子横向弛豫时间T2、溶胶的质子横向弛豫时间T2s、二阶矩的比率因子q及网链间分子量Mc.表中数据的误差值由数据拟合过程给出.分析表 2的数据,当硫化时间较小时,随着硫化时间的变大,网链间分子量Mc逐渐变小,交联链信号的比例A逐渐变大,交联部分和溶胶部分的质子横向弛豫时间T2和T2s逐渐变小.这是因为在硫化初期,交联点不断形成,交联密度逐渐变大,所以Mc逐渐变小,内部交联链信号比例A逐渐变大.由于交联点的形成限制了链段的运动,所以交联密度的增加降低了链段的运动性,从而导致交联部分和溶胶部分的质子横向弛豫时间T2、T2s变小.硫化到10 min以后,交联链信号比例A趋于稳定,悬尾链信号比例B变小,网链间分子量Mc变大.这是因为此时交联剂硫磺消耗完毕,交联反应停止,此时发生过硫反应,交联键出现断裂,交联密度开始变小.

|
图 5 橡胶样品的质子横向弛豫曲线,实验序列为Hahn回波脉冲序列 Figure 5 T2 decay curve of the rubber sample. The data was acquired using the Hahn echo pulse sequence |
| 表 2 1H Hahn回波法横向弛豫的拟合参数及计算的Mc Table 2 The parameters used in the 1H Hahn echo T2 fitting and the yielded Mc |
1H CPMG序列也是经常用来测量质子横向弛豫的一种脉冲序列.从脉冲序列的构成上来看,1H CPMG序列就是由一系列的1H Hahn回波组成.与1H Hahn回波相比,1H CPMG序列对磁场的不均匀性具有更好的会聚消除作用,因此在主磁场均匀性较差的低场NMR谱仪上应用较1H Hahn回波更为广泛.在本文的工作中,我们利用1H CPMG回波序列对硫化时间为3 min、7 min、10 min、30 min和60 min的橡胶进行了质子横向弛豫的测量.实验中使用的回波时间为0.1 ms,图 6以硫化3 min的胶料为例展示了1H CPMG回波测得的质子横向弛豫,并利用(1)式对曲线拟合,拟合得到的交联链的比例A、悬尾链的比例B、溶胶的比例C、交联链和悬尾链的弛豫时间T2、溶胶的弛豫时间T2s、二阶矩的比率因子q及网链间分子量Mc列于表 3.表中数据的误差值由数据拟合过程给出.分析表 3的数据,我们发现网链间分子量Mc随着硫化时间先变小后变大.和2.2节中1H Hahn回波测得的结果相比,1H CPMG序列测得的网链间分子量Mc要小,这是由于1H CPMG序列是由一系列回波时间相同的1H Hahn回波构成的,1H CPMG序列与1H Hahn法测得的质子横向弛豫不同.但是两种方法测得的交联密度变化趋势都是先变大后变小,这是因为1H CPMG序列与1H Hahn回波序列测得的质子横向弛豫都受到了残余偶极相互作用的调制,因此都在一定程度上反映交了体系交联密度的变化过程.

|
图 6 橡胶样品的质子横向弛豫曲线,实验序列为CPMG脉冲序列 Figure 6 T2 decay curve of the rubber sample. The data was acquired using the CPMG pulse sequence |
| 表 3 1H CPMG回波法横向弛豫的拟合参数及计算的Mc Table 3 The parameters used in the 1H CPMG T2 fitting and the yielded Mc |
下面对上述三种NMR方法测量的橡胶交联密度进行比较.为了理解材料中物理缠结和化学交联对NMR测试结果的贡献,我们还使用平衡溶胀法测量上述胶料样品的交联密度.实验中使用的溶剂为甲苯,表 4列出了溶胀前的质量w1、溶胀平衡时的质量w2、溶胀度Q及根据(8)式计算得到的网链间分子量Mc.随着硫化时间的增加,网链间分子量Mc同样呈现先变小后变大的趋势.
| 表 4 平衡溶胀法测量网链间分子量Mc Table 4 The Mc values measured using the equilibrium swelling approach |
表 5给出了4种不同方法测得的样品的交联密度.比较表 5的数据,我们发现不同方法之间测得的网链间分子量Mc差异较大,但是不同方法的测试结果都可以看到交联密度随硫化时间先变大后变小的趋势.
| 表 5 不同方法测量的Mc的比较 Table 5 Comparison of the Mc acquired using different approaches |
为了理解这些差异,我们对表 5中的数据进行了相关性分析.相关性分析是用来揭示变量之间是否存在相关性及相关密切程度的一种统计学方法,相关性系数r越接近1,说明两组数据的相关性越好.我们使用Excel中的Correl函数计算不同方法测试结果之间的相关性系数r,相关性系数的计算见(10)式,不同测试方法拟合结果之间的相关性系数见表 6.
| $ r={\rm{Correl}}(X, Y)=\frac{\Sigma (x-\bar{x})(y-\bar{y})}{\sqrt{\Sigma {{(x-\bar{x})}^{2}}\Sigma {{(y-\bar{y})}^{2}}}} $ | (10) |
| 表 6 不同方法的测得Mc的相关性系数 Table 6 Correlation efficient (r) of the Mc values obtained by different approaches |
1H DQ NMR与1H Hahn回波测试结果的相关性系数很高,为0.99,说明这两种方法测试结果的趋势一致性很高.这是因为1H DQ相干的激发效率取决于交联网络体系的残余偶极相互作用Dres,交联点使得链段运动受到限制,Dres变大.由于物理交联和化学交联都会使得体系的残余偶极相互作用增大,所以1H DQ NMR测得的是化学交联和物理交联的总和.残余偶极相互作用是横向弛豫的重要原因之一,因此1H Hahn回波通过测量质子横向弛豫而得到橡胶交联网络的信息也包括了化学交联和物理交联[21].虽然两种方法测得的交联密度值有差异,但都反映了化学交联和物理交联的总和.这可能是这两种方法测量值相关性系数很高的原因之一.
对于1H CPMG序列来说,虽然该方法也测量了橡胶的质子横向弛豫,但是测试结果与上述两种方法之间的相关性系数较低,都只有0.83(DQ-CPMG和Hahn-CPMG).我们认为这很可能与1H CPMG实验中选取的回波时间有关.有文献报道,在高分子体系中,1H CPMG序列测得的质子横向弛豫依赖于所使用的回波时间[31-33].这种对回波时间的依赖性与聚合物链段运动有关.聚合物中广泛存在的链段运动是一种热运动,其相关时间通常呈对数高斯分布.使用特定回波时间的1H CPMG序列对具有不同运动速率的高分子链段质子信号具有不同的回聚效果.因此,对于1H CPMG序列来说,使用特定回波时间可能意味着对材料中某一部分高分子链段质子信号的“选择性”观测.从这一点看来,与1H CPMG相比,在实验中使用不同回波时间的1H Hahn回波序列更能反映高分子材料中分子运动特点,因此更适合橡胶材料交联密度的测量[31, 32].对于1H DQ NMR来说,其理论模型是建立在高分子链段运动相关时间呈对数高斯分布这一基础之上的,所以,1H DQ NMR和1H Hahn回波的测量结果相关性高,而这两个方法的测量结果与1H CPMG的测量结果相关性较低.
平衡溶胀法与上述讨论的1H Hahn回波及1H DQ NMR的相关性系数分别为0.86和0.85.说明平衡溶胀法的测试结果与1H Hahn回波及1H DQ NMR的测试结果一致性较低.我们认为这主要是由于平衡溶胀法忽略了橡胶材料中广泛存在的物理缠结.在橡胶逐渐溶胀平衡的过程中,在溶剂分子的作用下橡胶中的交联网络逐步张开,同时部分分子间作用较弱的物理缠结则逐渐解开.因此,平衡溶胀法测得的为橡胶的化学交联密度及部分物理缠结密度的和.
综上所述,1H Hahn回波和1H DQ NMR测得的交联密度为物理交联和化学交联之和,平衡溶胀法测得是化学交联密度及部分物理缠结之和,而1H CPMG回波的测量值对回波时间有依赖性.
2.5 通过改进的1H CPMG序列测量橡胶交联密度在低场NMR谱仪上,由于磁体通常不配备匀场系统,典型的磁场不均匀性在100 Hz以上.过高的磁场不均匀性常常会导致1H DQ NMR与1H Hahn回波的测量误差.而1H CPMG序列可以很好的“消除”磁场不均匀性对1H横向弛豫带来的影响,因此比较适合在磁场均匀性差的低场NMR谱仪上使用.本节将探讨如何改进常规的1H CPMG序列,提高其测量值与1H Hahn回波测量值的相关性,使该序列能够在低场NMR谱仪上实现对橡胶样品交联密度的可靠测量.
根据上述讨论,我们推测1H CPMG实验中回波时间的选取是其相关性低的主要原因.为了证实这一推测,我们选取了0.05 ms、0.1 ms、0.2 ms和0.3 ms四个回波时间对橡胶样品进行测量.实验在VTMR20-010V-T型交联密度测试仪上进行,样品选取了硫化时间为3 min、7 min、10 min、30 min和60 min的胶料.图 7以硫化3 min胶料为例展示了不同回波时间时1H CPMG序列测量的质子横向弛豫,方框、圆点、正三角和倒三角的点分别代表回波时间为0.05 ms、0.1 ms、0.2 ms和0.3 ms时1H CPMG序列测量的质子横向弛豫曲线.分析图 7,我们发现回波时间不同时,1H CPMG序列测得的质子横向弛豫有所不同:回波时间越大,质子横向弛豫衰减的越快.

|
图 7 相同样品在不同回波时间时的质子横向弛豫曲线.脉冲序列为CPMG,回波时间(τ)的范围为0.05~0.3 ms Figure 7 T2 relaxation decay curves of the same sample with different echo times. The pulse sequence is CPMG. The echo times (τ) in the CPMG experiments were varied from 0.05~0.3 ms |
我们对不同回波时间下测得的横向弛豫曲线拟合得到了交联密度,结果见表 7.比较表 7的数据,我们发现回波时间不同时,1H CPMG序列测得的交联密度存在明显的差异.为了理解这些差异,我们将计算的结果与1H DQ NMR的测试结果进行相关性分析,相关性系数见表 7.由表中可知,当回波时间不同时,1H CPMG法测得的交联密度与1H DQ NMR之间的相关性不同.当回波时间为0.05 ms时,1H CPMG法与1H DQ NMR的测试结果相关性最低,只有0.69.回波时间为0.2 ms时,1H CPMG回波测得的结果与1H DQ NMR相关性最好,达到0.87.我们将四种回波时间的质子横向弛豫曲线相加,重新进行了归一化及拟合,相关计算结果见表 7.
| 表 7 不同回波时间时CPMG序列对同组橡胶测量Mc值的比较 Table 7 The Mc values of the rubber samples acquired by CPMG pulse sequence with different echo times |
表中数据的误差值由数据拟合过程给出.累加后的质子横向弛豫曲线包含了四种不同回波时间下采集的信号,测试结果与1H DQ NMR的相关系数为0.90,明显高于上述四种不同回波时间下的测试结果.沿着这一思路,我们认为通过合理设计1H CPMG序列中的回波时间,将有可能进一步提高1H CPMG测量值与1H DQ NMR测量值的相关性.我们实验室正在开展这一方面的工作.
3 结论本文对三种测量橡胶交联密度的NMR方法(1H DQ NMR、1H Hahn回波和1H CPMG回波)进行了较为系统地比较和讨论.探讨了这三种NMR方法在测量橡胶交联密度方面的差异,并讨论这些差异的来源.现将本文工作总结如下:
1、NMR技术测得的橡胶交联密度常常大于平衡溶胀法的测量值.其主要原因是,NMR测得的橡胶交联密度是材料中化学交联和物理交联之和,而平衡溶胀法测量得到的是橡胶材料的化学交联密度及部分物理缠结密度.
2、在三种NMR技术(1H DQ NMR、1H Hahn回波和1H CPMG回波)中,1H Hahn回波和1H DQ NMR的测量值具有很高的相关性.其原因在于这两种方法都考虑了高分子链段运动性分布对于测量值的影响.1H CPMG回波的测量值与上述两种方法的相关性不高,主要原因在于这一方法通常使用固定回波时间,无法准确反映高分子链段运动性分布对于测量值的影响.
3、1H CPMG回波法测量值对回波时间存在依赖性.通过叠加不同回波时间下1H CPMG弛豫数据可以有效地提高1H CPMG回波法测量值和1H Hahn回波/1H DQ NMR测量值的相关性.这一结果指出可以通过调节1H CPMG序列中的回波时间,以实现对橡胶样品中交联密度的可靠测量.
| [1] |
WANG Z L. Crosslinking density of rubbers and the method of determination[J].
World Rubber Industry, 1998, 25(4): 41-46.
王作龄. 橡胶的交联密度与测定方法[J]. 世界橡胶工业, 1998, 25(4): 41-46. |
| [2] |
ZHU R P, ZOU B, YANG J. Effect of crosslinking density on dynamic performance of natural rubber[J].
Special Purpose Rubber Products, 2009, 30(1): 43-45.
朱闰平, 邹波, 杨军. 交联密度对天然橡胶动态性能的影响[J]. 特种橡胶制品, 2009, 30(1): 43-45. |
| [3] | SCHLOGL S, TRUTSCHEL M L, CHASSÉ W, et al. Entanglement effects in elastomers:macroscopic vs microscopic properties[J]. Macromolecules, 2014, 47(9): 2759-2773. DOI: 10.1021/ma4026064. |
| [4] | HAVRÁNEK A, HEINRICH G. Experimental and theoretical analysis of viscoelastic behavior of different polymer networks[J]. Acta Polym, 1988, 39(10): 563-567. DOI: 10.1002/actp.1988.010391009. |
| [5] | VILGIS T A, HEINRICH G. The essential role of network topology in rubber elasticity[J]. Angew Makromol Chem, 2003, 202(1): 243-259. |
| [6] | VILGIS T A, HEINRICH G. New aspects in rubber elasticity:a challenge for theoretical physics and applied materials sciences[J]. Kautschuk Und Gummi Kunststoffe, 1992, 45(12): 1006-1013. |
| [7] | SCHIMMEL K H, HEINRICH G. The influence of the molecular weight distribution of network chains on the mechanical properties of polymer networks[J]. Colloid Polym Sci, 1991, 269(10): 1003-1012. DOI: 10.1007/BF00657430. |
| [8] | VILGIS T A, GERT H. Statics and dynamics of heterogeneous polymer networks[J]. Macromol Theor Simul, 1994, 3(3): 271-293. |
| [9] | MULLINS L. Determination of degree of crosslinking in natural rubber vulcanization. Part Ⅰ[J]. J Polym Sci, 1956, 19(92): 225-236. DOI: 10.1002/pol.1956.120199201. |
| [10] | GENNES P G D. Reptation of a polymer chain in the presence of fixed obstacles[J]. J Chem Phys, 1971, 55(2): 572-579. DOI: 10.1063/1.1675789. |
| [11] | KUHN W, PEREGI E, FEI Z, et al. Network dynamics of crosslinked polymers-crosslinking, filler and aging characterized by NMR parameters[J]. Mater Res Soc Symp Proc, 1991, 33(1): 217-223. |
| [12] | KUHN W, BARTH P, HAFNER S, et al. Material properties imaging of cross-linked polymers by NMR[J]. Macromolecules, 1994, 27(20): 5773-5779. DOI: 10.1021/ma00098a035. |
| [13] |
ZHAO F, BI W N, ZHANG P, et al. Influence of accelerators on structure of sulfur-cured natural rubber by nuclear magnetic resonance spectroscopy[J].
China Synthetic Rubber Industry, 2008, 31(1): 50-53.
赵菲, 毕薇娜, 张萍, 等. 用核磁共振法研究促进剂对硫磺硫化天然橡胶结构的影响[J]. 合成橡胶工业, 2008, 31(1): 50-53. |
| [14] | HAHN E L. Spin echoes[J]. Phys Rev, 1950, 80: 580-594. DOI: 10.1103/PhysRev.80.580. |
| [15] |
ZHAO S Y, WANG Y Y, ZHANG R R. Low-field NMR studies on the structure and dynamics of nanocomposite hydrogels[J].
Chinese J Magn Reson, 2014, 31(2): 172-184.
赵守远, 王媛媛, 张荣纯. 低场固体NMR研究纳米复合凝胶结构与动力学[J]. 波谱学杂志, 2014, 31(2): 172-184. |
| [16] | GAO Y, ZHANG R C, LV W F, et al. Critical effect of segmental dynamics in polybutadiene/clay nanocomposites characterized by solid state 1H NMR spectroscopy[J]. J Phys Chem C, 2014, 118(10): 5606-5614. DOI: 10.1021/jp5013472. |
| [17] | CARR H Y, PURCELL E M. Effect of diffusion on free precession in nuclear magnetic resonance experiments[J]. Phys Rev, 1954, 94(3): 630-638. DOI: 10.1103/PhysRev.94.630. |
| [18] | MEIBOOM S, GILL D. Modified spin-echo method for measuring nuclear relaxation times[J]. Rev Sci Instrum, 2004, 29(8): 688-691. |
| [19] | BAUM J, PINES A. NMR studies of clustering in solids[J]. J Am Chem Soc, 1986, 108(24): 7447-7454. DOI: 10.1021/ja00284a001. |
| [20] | HEUERT U, KNÖRGEN M, MENGE H, et al. New aspects of transversal 1H-NMR relaxation in natural rubber vulcanization[J]. Polym Bull, 1996, 37(4): 489-496. DOI: 10.1007/BF00556810. |
| [21] | SIMON G, BIRNSTIEL A, SCHIMMEL K H. Network characterization of end-linked poly(dimethyl siloxane) by 1H NMR spin-spin relaxation[J]. Polym Bull, 1989, 21(2): 235-241. |
| [22] | FRY C G, LIND A C. Determination of crosslink density in thermoset polymers by use of solid-state proton NMR techniques[J]. Macromolecules, 1988, 601(2): 921-929. |
| [23] | FETTERS L J, LOHSE D J, RICHTER D, et al. Connection between polymer molecular weight, density, chain dimensions, and melt viscoelastic properties[J]. Macromolecules, 1994, 27(17): 4639-4647. DOI: 10.1021/ma00095a001. |
| [24] | SAALWÄCHTER K, KLÜPPEL M, LUO H, et al. Chain order in filled SBR elastomers:a proton multiple-quantum NMR study[J]. Appl Magn Reson, 2004, 27(3, 4): 401-417. |
| [25] | SAALWÄCHTER K, ZIEGLER P, SPYCKERELLE O, et al. 1H multiple-quantum nuclear magnetic resonance investigations of molecular order distributions in poly (dimethyl siloxane) networks:Evidence for a linear mixing law in bimodal systems[J]. J Chem Phys, 2003, 119(6): 3468-3482. DOI: 10.1063/1.1589000. |
| [26] | SAALWÄCHTER K. 1H multiple-quantum nuclear magnetic resonance investigations of molecular order in polymer networks. Ⅱ. Intensity decay and restricted slow dynamics[J]. J Chem Phys, 2004, 120(1): 454-64. DOI: 10.1063/1.1630561. |
| [27] | SAALWÄCHTER K, AND B H, LÓPEZMANCHADO M A. Chain order and cross-link density of elastomers as investigated by proton multiple-quantum NMR[J]. Macromolecules, 2005, 38(38): 9650-9660. |
| [28] | GRAF R, HEUER A, SPIESS H W. Chain-order effects in polymer melts probed by 1H double-quantum NMR spectroscopy[J]. Phys Rev Lett, 1998, 80(26): 5738-5741. DOI: 10.1103/PhysRevLett.80.5738. |
| [29] | FLORY P J., REHNER J. Statistical mechanics of cross-linked polymer networks Ⅱ Swelling[J]. J Chem Phys, 1943, 11: 521-526. DOI: 10.1063/1.1723792. |
| [30] | PATEL S K, MALONE S, COHEN C, et al. Elastic modulus and equilibrium swelling of poly(dimethyl siloxane) networks[J]. Macromolecules, 1992, 25: 5241-5251. DOI: 10.1021/ma00046a021. |
| [31] | BREMNER T, RUDIN A. The effect of degassing on the measurement of transverse relaxation times in molten polymers[J]. J Polym Sci Poly Phys, 1996, 34(11): 1893-1902. DOI: 10.1002/(ISSN)1099-0488. |
| [32] | LITVINOV V M, RIES M E, BAUGHMAN T W, et al. Chain entanglements in polyethylene melts. Why is it studied again[J]. Macromolecules, 2013, 16(2): 541-547. |
| [33] | WHITTAKER A K, BREMNER T, ZELAYA F O. The effect of field inhomogeneous and molecular diffusion on the NMR transverse relaxation behavior of polymer melts[J]. Polymer, 1995, 36(11): 2159-2164. DOI: 10.1016/0032-3861(95)95291-8. |