 2016, Vol. 33
2016, Vol. 33文章信息
- 杨贤鹏, 俞豪杰, 王立
- YANG Xian-peng, YU Hao-jie, WANG Li
- 二茂铁基化合物的ESR研究进展
- Recent Progresses in ESR Studies on Ferrocenyl Compounds
- 波谱学杂志, 2016, 33(3): 491-501
- Chinese Journal of Magnetic Resonance, 2016, 33(3): 491-501
- http://dx.doi.org/10.11938/cjmr20160314
-
文章历史
收稿日期: 2015-08-20
收修改稿日期: 2016-07-08




DOI:10.11938/cjmr20160314


二茂铁[Fe(C5H5)2,Fc]具有π键平面配体夹心结构(Sandwich):两个平行的环戊二烯基负离子处于两边,一个铁(Ⅱ)离子处于中间,茂环上所有的碳原子都与铁原子成键,C-Fe键键长相等.二茂铁基化合物因具有独特的氧化还原、电、光、磁和催化等性能而被广泛研究与应用.它最显著的特点是氧化还原可逆性,即二价铁与三价铁在不同的氧化还原条件下可以相互转化: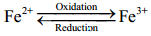
二茂铁离子(Fc+)的电子自旋通常伴随着快速自旋-晶格弛豫(T1)[3],需在低温条件下获得ESR信号,与自由电子的g因子(ge)相差较大.根据测试条件及g因子值的大小等可以来判断ESR信号的来源[4],进而研究二茂铁基化合物电子结构、自旋密度、分子间及分子内相互作用等.
在早期研究工作中,研究者较多地关注二茂铁离子、取代二茂铁离子以及联二茂铁离子的ESR信号[5].二茂铁络合物兼具σ-π配位结构,受当时条件限制,所得分子轨道[6]不是非常精确(图 1),但揭示了二茂铁的电子结构,将二茂铁离子的ESR信号与各分子结构参数直观联系起来.Prins等人[7]最早成功地用ESR技术研究Fc+.在此之前,有人在78 K温度下进行了尝试,但并没有得到Fc+的信号,因为Fc+的电子组态为2E2g[(a1g)2(e2g)3],波函数中自旋-轨道和轨道角动量算符远偏离于0,T1非常短.在更低的温度下,由于弛豫时间变长,才有可能观察到Fc+的ESR信号.Prins等[7]在20 K条件下观察到了四氟硼酸二茂铁(Fc+·BF4-)和二茂铁三碘化物(Fc+·I3-)在二甲基甲酰胺(DMF)和丙酮中的固溶体的ESR信号;而在20 K与78 K下均可以观察到苯基二茂铁离子(PhFc+)和1, 1’-二苯基二茂铁离子(DPFc+),只是78 K下其信号变宽变弱.Duggan[8]则用ESR技术探测了不同取代基二茂铁离子的电子结构,包括十甲基二茂铁离子(DecaMFc+)、1, 1’-二甲基二茂铁离子(DMFc+)和1, 1’-三甲烯二茂铁离子(TMFc+),结果表明分子的低对称扭曲(low-symmetry distortions)越大,g因子越接近于2.0,二茂铁离子对称性越低(取代基、抗衡离子的影响),能级分裂越高.联二茂铁阳离子的ESR信号可在77 K下得到,由于取代基和抗衡离子的影响,ESR信号呈各向异性.Prins和Duggan等[7-9]测得了大量二茂铁基化合物的g因子(如表 1所示).g因子值远偏离自由电子的2.002 3,表明轨道对g因子的贡献大;二茂铁离子g因子与分子的低对称扭曲程度和晶体场强度相关联;取代基处于s骨架上,对p没有直接影响,不会显著影响3d2与e2g之间p配位的强度;对称扭曲参数比e1g*和e2g轨道之间配位场分裂小1或2个数量级,取代二茂铁离子可近似作一级轴对称处理.另外Prins[6, 8]还给出了二茂铁离子的g因子表达式:
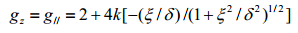 (1)
(1)
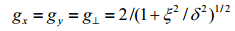 (2)
(2)

| Compound | g∥ | g⊥ | k | δ**/cm–1 |
| 二茂铁离子(Fc+) | 4.35 | 1.26 | 0.76 | 270 |
| 甲基二茂铁离子(MeFc+) | 4.17 | 1.47 | 0.83 | 360 |
| 苯基二茂铁离子(PhFc+) | 3.98 | 1.58 | 0.80 | 420 |
| 1, 1’-二甲基二茂铁离子(DMFc+) | 3.83 | 1.67 | 0.83 | 500 |
| 1, 1’-二苯基二茂铁离子(DPFc+) | 3.63 | 1.74 | 0.82 | 580 |
| 1, 1’-二丁基二茂铁离子(DBFc+) | 3.88 | 1.68 | 0.86 | 500 |
| 苯基羰基二茂铁离子(PCFc+) | 3.69 | 1.73 | 0.84 | 570 |
| 乙酰基二茂铁离子(AcFc+) | 3.62 | 1.76 | 0.85 | 610 |
| 1, 1’-二甲基二茂铁离子(DMFc+·PF6–) | 4.00 | 1.92 | / | 1350 |
| 1, 1’-二甲基二茂铁离子(DMFc+·TCA–·TCAA) | 4.44 | 1.22 | / | 310 |
| 1, 1’-三甲烯二茂铁离子(TMFc+·PF6–) | 3.86 | 1.81 | / | 850 |
| 1, 1’-三甲烯二茂铁离子(TMFc+·TCA–·2TCAA) | 3.83 | 1.64 | / | 575 |
| 十甲基二茂铁离子(DecaMFc+·PF6–) | 4.43 | 1.35 | / | 365 |
| 十甲基二茂铁离子(DecaMFc+·TCA–·2TCAA) | 4.37 | 1.26 | / | 324 |
| *k为轨道折减系数;**d为晶体场强度. | ||||
k为轨道折损系数,δ为晶体场强度,ξ为自旋轨道耦合常数.通常Δg=g∥-g⊥ < 0.8,表明可离域或者电子转移速度高于ESR的时间尺度(109~1010 s-1)[10].用g因子可计算有效磁矩(effective magnetic moments)与分子的扭曲因子(distortion factors)[11].
早期的探索不仅在确定二茂铁基电子结构上起到重要作用,其积累的大量数据也为之后的研究奠定了基础.近年来也有联二茂铁的ESR研究,与早期研究的区别在于近期的研究更加注重功能化与应用.Lohan等人[12]合成了1’, 1’’’-双乙炔基联二茂铁,并将之作为金、钌和锇的连接基团.在此之前,混合价化合物1’, 1’’’-双取代联二茂铁离子的电子转移速率与g张量各向异性(g-tensor anisotropy)之间的关系已经建立起来[5, 13, 14].陷阱态化合物(trapped mixed-valence complexes)的电子转移速率小于108 s-1,各向异性较大(Δg > 1.5);去陷阱态化合物(detrapped mixed-valence complexes)的电子转移速率大于108 s-1,各向异性较小(Δg < 1.1).因此,根据测得的Δg可知两个二茂铁基团的电子转移速率以及自旋相互作用的情况.此外还可以根据ESR谱图的轴对称性来判断未成对电子不在钌离子/锇离子上,而在有明显联二茂铁特征的单占分子轨道(SOMOs)上.
自旋密度对于二茂铁基化合物的ESR研究非常重要.Rao等人[15]合成了三甲苯基硼修饰的苯并咪唑基二茂铁离子(化合物1+),并用ESR探究了分子的自旋离域[图 2(a)].结果发现ESR信号呈高度各向异性(g∥=3.11,g⊥=1.94),其远小于无取代的Fc+(g∥≈3.80),这表明自旋密度部分延伸到有机取代基团,进而说明Fc+中存在三价铁到苯基基团的自旋离域.Kusamoto等人[16]研究了二茂铁-二硫纶([FcS4dt(Me)2]•+)间强供体-受体之间的电子相互作用,[FcS4dt(Me)2]•+(化合物2•+)在7.5 K下的ESR谱如图 2(b)所示,g∥=3.70、g⊥=1.70,表明其ESR信号呈高度各向异性.而二茂铁盐衍生物的各向异性及相关参数(表 1)表明衍生物的自旋密度位于二茂铁基团的铁原子上.[FcS4dt(Me)2]•+的g因子更小,说明自旋密度扩展到了共轭的二硫纶骨架上,[FcS4dt(Me)2]•+的最高占据分子轨道(HOMOs)主要位于二茂铁基团,部分在π-共轭的二硫纶上,这与CV、DFT模拟结果相符.图 2(b)中300~400 mT之间较小的信号来自于分解的基团.

ESR在表征二茂铁基混合价化合物上也有所应用.Siebler等人[10]合成联二茂铁氨基酸,用碘氧化后,得到的ESR谱图中的Δg为1.08和1.09,这表明存在混合价态的二茂铁离子.Sixt等人[17]则通过ESR图谱来判断二茂铁基化合物氧化过程的氧化部位.二价的钌离子与二茂铁中亚铁离子都易被氧化,两者氧化后的ESR信号差异很大,由此可用来判断氧化部位.
2 二茂铁基化合物分子间作用探测分子间相互作用不同导致分子的外部环境不同,通过ESR的g因子或者超精细耦合常数(hfc)的信息,可以探测分子间的相互作用.
Fc在MCM-41孔道中氧化[18],得到的ESR信号(g∥=4.32±0.03,g⊥=2.06±0.02)与Fe+·I3-中Fc+的ESR信号(g∥=4.36,g⊥=1.30)[19]相比,g⊥相差较大,这是因为Fc在MCM-41孔道中易被氧化,所得Fc+在硅孔道中稳定,两个体系分子间相互作用不同.此处作者并没有对氧化机理进行探讨,但根据样品处理过程中的现象(颜色变化)和MCM-41负载Fc后的洗涤过程可能接触氧气,推测具体氧化机理为:
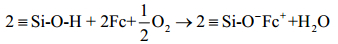
该机理后来被Schnitzler等人[20]的研究所验证.Schnitzler等人[20]将Fc负载到多孔石英玻璃上,证明了Fc的氧化过程发生在负载之后,且需要氧气.ESR结果与Toda等人[18]的研究类似,并且发现Fe3+存在不同的形态:高自旋、斜方与立方结构.Wang等人[21]利用ESR技术发现二茂铁硅烷可与四腈基乙烯形成络合物,环戊二烯基作为电子供体,乙烯基作为电子受体,两者之间p轨道的重叠发挥着重要作用[22-24].Yan等人[25]制备了二茂铁基多金属氧簇超分子复合物,该复合物的ESR信号与二茂铁基和四氰基乙烯形成的电荷转移络合物类似,二茂铁基与多金属氧簇形成的也是电荷转移络合物.与二茂铁离子不同,该复合物可在常温下产生未成对电子,因此有常温下的ESR信号.Fukuzumi等人[26]则利用二茂铁基化合物形成络合物前后ESR信号的差别来证明离子络合物的形成.
Song等人[27]制备了聚丙二醇(PPG)、Fc、β-环糊精(β-CD)三元聚集体(ternary aggregates),并用X光电子能谱(XPS)与ESR测定Fe(ii)的电子结构,来研究聚集体结构的变化与Fe(Ⅱ)电子结构变化之间的关系.游离二茂铁与它的两个聚集体的ESR谱如图 3所示:ESR信号强度随二茂铁组分的减少而减弱;对于三元聚集体,二茂铁含量减少,ESR信号的对称性亦减弱.三个信号的g因子分别为2.12(ga)、2.25(gb)和2.02(gc),而且聚集体之间的g因子差别大于纯二茂铁与聚集体之间的差别,这是由聚集体之间的分子间相互作用不同致使Fe(Ⅱ)外部环境不同,进而使Fe(Ⅱ)的价电子密度不同造成的.但该结果与公认的二茂铁的ESR结果不一致,一般认为还原态二茂铁及常温下氧化态二茂铁离子没有ESR信号,低温下二茂铁离子的ESR信号呈各向异性.二者之间不一致可能有两种原因:一是原料二茂铁没有经过纯化处理,采集到杂质信号,如果是这样,杂质充当了自旋探针的作用;二是因为二茂铁与环糊精形成主客体,它们相互作用造成电子结构变化,但至今还未见这方面的报道,难以下结论.

上述例子均利用ESR技术来探测分子间相互作用,或证实分子间作用的存在,或表明不同分子间相互作用的区别.
3 二茂铁基化合物分子内作用探测ESR可以用来探测基团间的电子相互作用,例如对环戊二烯基·环庚三烯基钒(trovacenyl)衍生物,用ESR可以获得分子内金属-金属相互作用的信息[28].而当作为两个trovacenyl的连接基团[化合物5,图 4(a)],二茂铁基是否存在电子相互作用,也可以通过ESR来证实[29].ESR结果显示一个钒原子核孤单电子(化合物5••)超精细耦合产生8-线谱图[8-line pattern,图 4(b)],而非基于2nI+1规则的15线谱图(15-line pattern).5••的超精细耦合值[(a(51V)=7.11 mT)]小于5, 5-双(环戊二烯基·环庚三烯基钒)二茂铁([5, 5]bitrovacene)的超精细耦合值(7.22 mT),而大于trovacene的超精细耦合值(6.98 mT).ESR结果说明二茂铁基是还原态的、抗磁性的p体系,所以其嵌入造成了|J| < < |a|、在ESR时间尺度内,两个trovacenyl基团在各自的金属中心离域,分子内金属-金属相互作用被二茂铁基团阻隔而大大减弱.而且两个顺磁中心之间的交换作用随着连接彼此间的抗磁性配体长度的增长而变弱.

|
| 图 4 (a)化合物5的合成;(b)5••的ESR谱图 Fig. 4 (a) Synthesis of compound 5. (b) X-band ESR spectrum of 5•• |
Chaicharoenwimolkul等人[30]合成了苯胺-二茂铁基苯胺共聚物[图 5(a)],并用ESR研究了二茂铁基团对聚合物顺磁特性的影响.共聚物的ESR谱图中[图 5(b)]显示峰强度随着二茂铁基的浓度增大而减弱,峰型变宽,不对称性增强.与聚苯胺的g因子(2.003 1)相比,当单体间二茂铁基苯胺与苯胺的摩尔比分别为5%、10%、15%和20%时,共聚物的g因子分别为2.003 2、2.003 5、2.003 8和2.003 9.从g因子的变化看,二茂铁基基本上不影响聚合物的顺磁性.峰强度的减弱可能是聚合物的共轭结构被扰乱与破坏引起的.作者还对上述共聚物进行二茂铁离子掺杂,并发现所有掺杂共聚物的g因子都为2.003 4,接近自由电子的值.

|
| 图 5 (a)聚(苯胺-co-二茂铁基苯胺)的制备;(b)不同二茂铁基苯胺对苯胺摩尔比的聚(苯胺-co-二茂铁基苯胺)的ESR图:二茂铁基苯胺的浓度分别为0(线1)、5%(线2)、10%(线3)、15%(线4)、20%(线5)、(聚二茂铁基苯胺)(线6) Fig. 5 (a) The preparation of poly (aniline-co-m-ferrocenylaniline). (b) ESR spectra of poly (aniline-co-m-ferrocenylaniline) (s) for 0(line 1), 5%(line 2), 10%(line 3), 15%(line 4), 20%(line 5) molar ratio of m-ferrocenylaniline to aniline, and poly (m-ferrocenylaniline) (line 6) |
许多二茂铁衍生物的取代基可形成稳定的自由基[31, 32],用ESR可以探测二茂铁衍生物取代基上自由基的信息.二茂铁基的两个环状配体可绕轴旋转,处于动态平衡.分子处于不同的氧化还原状态下,平衡方向会有所移动,ESR信号会发生变化.因此,在特定条件下可通过ESR技术来判断二茂铁基化合物分子所处的构象.Takai等人[33]构建了7分子(二茂铁-萘二胺共轭体系,Fc-NDIs)[图 6(a)],结果显示分子绕二茂铁基纵轴旋转受温度与电子转移的影响.在中性298 K条件下,NDIs基团快速旋转.当处于1电子还原态(oneelectron reduced state)时,由于两个NDIs基团之间的π键相互作用,[7]•-构象为关闭状态[图 6(e)].进一步还原之后,[7]2-则处于关闭-开启平衡状态.在相应的ESR谱图中可以看到,相比于[7]•-,[7]2-峰强度大为减弱,根据NDI•-与TDAE•+的强度比,可计算出顺磁构象与反磁构象所占比例分别为21%与79% [图 6(b)].逐步降低温度,[7]2-的ESR信号进一步减弱,顺磁构象减少[图 6(c)],两个构象的平衡常数与温度的关系符合范特霍夫方程[图 6(d)].

|
| 图 6 (a) 7分子(二茂铁-萘二胺共轭体系,Fc-NDIs)的开启形式(左)和关闭形式(右);(b) [7]•-(线1)还原成[7]2-(线2)的ESR信号变化,星号表示Mn2+标记;(c) [7]2-随温度变化的ESR信号;(d) [7]2-顺磁构象和反磁构象的平衡常数-温度的范特霍夫方程拟合;(e) 7分子氧化还原控制的绕轴旋转 Fig. 6 (a) The open form (left) and the closed form (right) of 7; (b) ESR spectral change in the course of the conversion from[7]•-(line 1) to [7]2- (line 2). Asterisks denote Mn2+ markers; (c) Temperature-dependent ESR spectral changes of [7]2-; (d) A van't Hoff plot for the formation constant of the closed form of [7]2-; (e) Description of redox-controlled pivoting motions of 7 |
Iordache等人[34]通过对比二茂铁单取代与双取代化合物的ESR图谱,发现单取代为顺磁性,而双取代为反磁性,从而进一步证明双取代化合物的π-二聚体结构.
也有将自旋捕捉ESR技术用于探测二茂铁基化合物的催化过程.Erben等人[35]将二茂铁基化合物用作醇酸树脂涂料的干燥剂,并用自旋捕捉ESR技术证明了氢过氧化物分解过程中Fc+的重要作用.过氧自由基与烷氧自由基会出现在干燥过程中,与自旋捕捉剂生成自旋加合物,产生ESR信号.
5 二茂铁基化合物结构研究中的密度泛函理论二茂铁基化合物的ESR表征中经常使用模拟技术.密度泛函理论(density functional theory, DFT)作为量子力学方法,可以用来计算多电子体系的电子结构,在NMR测试中化学位移与ESR测试中g-张量(g-tensor)的计算中有着广泛的应用[36].研究中经常采取实验与计算结合的方式,通过对比实验值与模拟计算值来获得比较可信的结果.例如ESR结果显示一价锰比二茂铁基更易被氧化,DFT计算进一步证实了这一结论[37].众多研究表明作二茂铁基化合物的ESR研究中,实验值与计算值相互验证发挥着越来越重要的作用.
6 二茂铁基化合物研究中ESR技术与其它技术的联用运用ESR技术可以有效探测二茂铁基化合物的结构,而ESR与其它多种技术的联用可以进一步验证结果的准确性.ESR技术可以与XPS、X光衍射(XRD)、UV-Vis spectra、红外光谱(FTIR spectra)、NMR、CV和穆斯堡尔谱(M ssbauer spectra)等多种测试手段一起使用来探测二茂铁基化合物的结构.例如用XPS和ESR可共同来探测配合态二茂铁基的电子结构[27];在Fc与Fc+转换过程研究中,除ESR外,还经常用NMR[30]与CV[30]来探测氧化还原的过程;用紫外-可见-近红外吸收光谱可以表征氧化前后电子结构的差异[15];穆斯堡尔谱与ESR时间尺度不同,一起使用可以测定电子转移速度[10];另外也可以利用二价铁与三价铁的穆斯堡尔谱的差异来计算被氧化的百分比[18].
7 展望二茂铁基化合物研究涉及配位化学、高分子化学、超分子化学、分析化学和有机化学等诸多化学领域,还与生物化学、生命科学等学科有交叉.二茂铁基化合物的研究是催化、生物传感器和分子器件等方面应用研究的热点之一,应给予关注.运用ESR技术开展这方面的研究可获得其它技术得不到的信息,合理科学地将ESR技术与其他表征手段结合起来,将有利于推进二茂铁基化合物的研究与应用.
| [1] | 杨茵, 陈家良, 苏循成, 等. 蛋白质顺磁标记技术与生物核磁共振中的赝接触位移[J]. 波谱学杂志 , 2014, 31 (2) : 155-171 Yang Yin, Chen Jia-liang, Su Xun-cheng, et al. Paramagnetic labeling of proteins and pseudocontact shift in structural biology[J]. Chinese J Magn Reson , 2014, 31 (2) : 155-171 |
| [2] | 赵保路. 利用电子自旋共振(ESR)技术研究一氧化氮自由基在心脑血管疾病和健康中的"双刃剑"作用[J]. 波谱学杂志 , 2015, 32 (2) : 195-207 Zhao Bao-lu. "Double edge" effects of nitric oxide free radical in cardio-brain-vascular diseases and health studied by ESR[J]. Chinese J Magn Reson , 2015, 32 (2) : 195-207 |
| [3] | Elschenbroich C, Bilger E, Ernst R D, et al. Closed, half open, and open ferrocenes redox behavior and electron spin resonance of the radical cations[J]. Organometallics , 1985, 4 (11) : 2 068-2 071 DOI:10.1021/om00130a029 |
| [4] | Jana R, Mobin S M, Schwederski B, et al. Variable coordination of redox-active TCNB in discrete and polymeric ferrocenylcopper(I) complexes: Structures and spectroelectrochemical behaviour[J]. Dalton Trans , 2013, 42 (45) : 16 142-16 150 DOI:10.1039/c3dt51360b |
| [5] | Dong T Y, Hendrickson D N, Pierpont C G, et al. Mixed-valence 1', 6'-Dihalobiferrocenium salts-the effect of the solid-state environment on electron-transfer rates[J]. J Am Chem Soc , 1986, 108 (5) : 963-971 DOI:10.1021/ja00265a021 |
| [6] | Prins R. Electronic structure of the ferricenium cation: Electron spin resonance measurements of the cations of ferrocene derivatives[J]. Mol Phys , 1970, 19 (5) : 603-620 DOI:10.1080/00268977000101641 |
| [7] | Prins R, Reinders F J. Electron spin resonance of the cation of ferrocene[J]. J Am Chem Soc , 1969, 91 (17) : 4 929-4 931 DOI:10.1021/ja01045a063 |
| [8] | Duggan D M, Hendrickson D N. Electronic structure of various ferricenium systems as inferred from raman, infrared, low-temperature electronic absorption, and electron paramagnetic resonance measurements[J]. Inorg Chem , 1975, 15 (4) : 955-970 |
| [9] | Prins R, Korswagen A R. Substituent effects in the ESR spectra of ferricenium cations[J]. J Organomet Chem , 1970, 25 (2) : C74-C76 DOI:10.1016/S0022-328X(00)87818-3 |
| [10] | Siebler D, Gasi T, Heinze K. Biferrocene amino acid, a ferrocenylogoue of ferrocene amino acid: Synthesis, cross-linking, and redox chemistry[J]. Organometallics , 2011, 30 (2) : 313-327 DOI:10.1021/om1010808 |
| [11] | Cowan D O, Candela G A, Kaufman F. Organic solid state. V. symmetry distortions in ferrocenium compounds[J]. J Am Chem Soc , 1971, 93 (16) : 3 889-3 893 DOI:10.1021/ja00745a011 |
| [12] | Lohan M, Ecorchard P, Lapinte C, et al. 1', 1'''-Bis(ethynyl) biferrocene as a linking group for gold, ruthenium, and osmium fragments: Synthesis, solid state structures, and electrochemical, UV-Vis, and EPR spectroscopical studies[J]. Organometallics , 2009, 28 (6) : 1 878-1 890 DOI:10.1021/om8011344 |
| [13] | Dong T Y, Schei C C, Hwang M Y, et al. Pronounced effect of substituents on the intramolecular electron-transfer rates in mixed-valence biferrocenium triiodide complexes[J]. Organometallics , 1992, 11 (2) : 573-582 DOI:10.1021/om00038a012 |
| [14] | Dong T, Chang L, Lee G, et al. Pronounced effects of zero-point energy difference on intramolecular[J]. Organometallics , 2002, 21 (20) : 4 192-4 200 DOI:10.1021/om0203476 |
| [15] | Rao Y, Kusamoto T, Sakamoto R, et al. Reactivity and electronic properties of a ferrocene molecule bearing an N, C-chelated BMes[J]. Organometallics , 2014, 33 (7) : 1 787-1 793 DOI:10.1021/om500138f |
| [16] | Kusamoto T, Takada K, Sakamoto R, et al. Ferrocene-dithiolene hybrids: Control of strong donor-acceptor electronic communication to reverse the charge transfer direction[J]. Inorg Chem , 2012, 51 (22) : 12 102-12 113 DOI:10.1021/ic300581a |
| [17] | Sixt T, Sieger M, Krafft M J, et al. Ambi-valence taken literally: Ruthenium vs iron oxidation in (1, 1'-diphosphinoferrocene) ruthenium(II) hydride and chloride complexes as deduced from spectroelectrochemistry of the heterodimetallic "mixed-valent" intermediates[J]. Organometallics , 2010, 29 (21) : 5 511-5 516 DOI:10.1021/om1004258 |
| [18] | Toda Y, Ishimaru S I, Ikeda R, et al. Oxidation of ferrocene molecules adsorbed in MCM-41[J]. J Phys Chem Solids , 2004, 65 (2, 3) : 471-473 |
| [19] | Ward H R, Lawler R G, Loken H Y, et al. Nuclear polarization in the products of chemical reactions occurring in the absence of a magnetic field[J]. J Am Chem Soc , 1969, 91 (17) : 4 928-4 929 DOI:10.1021/ja01045a062 |
| [20] | Schnitzler M C, Mangrich A S, Macedo W A A, et al. Incorporation, oxidation and pyrolysis of ferrocene into porous silica glass: a route to different silica/carbon and silica/iron oxide nanocomposites[J]. Inorg Chem , 2006, 45 (26) : 10 642-10 650 DOI:10.1021/ic061312r |
| [21] | Wang L, Wang X J, Pan J, et al. Study on ESR spectra of poly(ferrocenyldimethylsilane)/TCNE and spin-probed poly[J]. J Appl Polym Sci , 2002, 86 (14) : 3 508-3 511 DOI:10.1002/(ISSN)1097-4628 |
| [22] | Tanaka H, Mizota K. Generation of a spin polymer through a charge-transfer complex of a ferrocenyl mesogen with tetracyanoethylene in the solid state[J]. Macromol Rapid Comm , 1995, 16 (1) : 1-7 DOI:10.1002/marc.1995.030160101 |
| [23] | Kuroda H. Polarized absorption spectra of single crystals of ferrocene and its molecular complexes[J]. J Mol Spectrosc , 1969, 30 (1-3) : 355-364 DOI:10.1016/0022-2852(69)90273-2 |
| [24] | Adman E, Rosenblum M, Sullivan S, et al. Structure of the ferrocene-tetracyanoethylene complex[J]. J Am Chem Soc , 1967, 17 (89) : 4 540-4 542 |
| [25] | Yan Y, Li B, He Q, et al. Synthesis and redox-responsive self-assembly of ferrocene grafted anderson-type polyoxometalate hybrid complexes[J]. Soft Matter , 2012, 8 (5) : 1 593-1 600 DOI:10.1039/C1SM06610B |
| [26] | Fukuzumi S, Okamoto K, Imahori H. Thermal intramolecular electron transfer in a ferrocene-naphthoquinone linked dyad promoted by metal ions[J]. Angew Chem Int Ed , 2002, 41 (4) : 620-622 DOI:10.1002/(ISSN)1521-3773 |
| [27] | Song L X, Du F Y, Yang J, et al. Fc-content dependence of composition, structure and degradation degree in supramolecular aggregates of polypropylene glycol, ferrocene and β-cyclodextrin[J]. Soft Matter , 2011, 7 (14) : 6 671-6 677 DOI:10.1039/c1sm05484h |
| [28] | Elschenbroich C, Plackmeyer J, Nowotny M, et al. Electro-and magnetocommunication in [5, 5]ditrovacenyls, [(η7-C7H7)V(η5-C5H4-X-η5-C5H4)V(η7-C7H7)], Mediated by the spacers X=(Z)-CH=CH-, (E)-CH=CH-, > C=CH2, -CH2CH2, and-CH2-**[J]. Chem Eur J , 2005, 11 (24) : 7 427-7 439 DOI:10.1002/(ISSN)1521-3765 |
| [29] | Elschenbroich C, Lu F, Harms K, et al. Electrochemical behavior and EPR study of the paramagnetic termetallocene Di([5]trovacenyl-tetramethyl-η5-cyclopentandienyl)iron[J]. Polyhedron , 2014, 79 : 300-305 DOI:10.1016/j.poly.2014.04.065 |
| [30] | Chaicharoenwimolkul L, Chairam S, Namkajorn M, et al. Effect of ferrocene substituents and ferricinium additive on the properties of polyaniline derivatives and catalytic activities of palladium-doped poly(m-ferrocenylaniline)-catalyzed suzuki-miyaura cross-coupling reactions[J]. J Appl Polym Sci , 2013, 130 (3) : 1 |
| [31] | Souto M, Morales D C, Guasch J, et al. Intramolecular electron transfer and charge delocalization in bistable donor-acceptor systems based on perchlorotriphenylmethyl radicals linked to ferrocene and tetrathiafulvalene units[J]. J Phys Org Chem , 2014, 27 (6) : 465-469 DOI:10.1002/poc.v27.6 |
| [32] | Ratera I, Ruiz-Molina D, Wurst K, et al. A new photomagnetic molecular system based on photoinduced self-assembly of radicals[J]. Angew Chem Int Ed , 2001, 40 (5) : 919-922 DOI:10.1002/(ISSN)1521-3773 |
| [33] | Takai A, Yasuda T, Ishizuka T, et al. A directly linked ferrocene-naphthalenediimide conjugate: Precise control of stacking structures of π-systems by redox stimuli[J]. Angew Chem Int Ed , 2013, 52 (35) : 9 167-9 171 DOI:10.1002/anie.201302587 |
| [34] | Iordache A, Oltean M, Milet A, et al. Redox control of rotary motions in ferrocene-based elemental ball bearings[J]. J Am Chem Soc , 2012, 134 (5) : 2 653-2 671 DOI:10.1021/ja209766e |
| [35] | Erben M, Veselý D, Vinklárek J, et al. Acyl-substituted ferrocenes as driers for solvent-borne alkyd paints[J]. J Mol Catal A-Chem , 2012, 353, 354 : 13-21 DOI:10.1016/j.molcata.2011.10.024 |
| [36] | Schreckenbach G, Ziegler T. Density functional calculations of NMR chemical shifts and ESR g-tensors[J]. Theor Chem Acc , 1998, 99 (2) : 71-82 DOI:10.1007/s002140050306 |
| [37] | Bezuidenhout D I, van der Westhuizen B, Swarts P J, et al. Redox behaviour of cymantrene fischer carbene complexes in designing organometallic multi-tags[J]. Chem Eur J , 2014, 20 (17) : 4 974-4 985 DOI:10.1002/chem.201304711 |