 2016, Vol. 33
2016, Vol. 33文章信息
- 宋本腾, 褚月英, 王吉清, 郑安民, 邓风
- SONG Ben-teng, CHU Yue-ying, WANG Ji-qing, ZHENG An-min, DENG Feng
- 分子间相互作用对核酸碱基中17O屏蔽张量与四极耦合常数影响的理论计算研究
- Influences of Intermolecular Interactions on the 17O Nuclear Magnetic Parameters in Nucleic Acid Bases: A Theoretical Investigation
- 波谱学杂志, 2016, 33(3): 378-394
- Chinese Journal of Magnetic Resonance, 2016, 33(3): 378-394
- http://dx.doi.org/10.11938/cjmr20160303
-
文章历史
收稿日期: 2015-09-22
收修改稿日期: 2016-07-12




DOI:10.11938/cjmr20160303


2. 波谱与原子分子国家重点实验室(中国科学院 武汉物理与数学研究所), 湖北 武汉 430071
2. State Key Laboratory of Magnetic Resonance and Atomic and Molecular Physics (Wuhan Institute of Physics and Mathematics, Chinese Academy of Sciences), Wuhan 430071, China
氢键广泛存在于生物体系中,是决定生物大分子三维结构的重要相互作用之一,与DNA和RNA等生物大分子的结构和功能有着密切的联系[1-4].核酸碱基是DNA和RNA的重要组成部分,因此,对核酸碱基结构的正确解析将有助于人们对DNA和RNA在遗传信息的传递以及生命活动中所扮演的角色有更深入的理解.
众所周知,核磁共振(NMR)是表征物质结构的重要技术手段之一,在生物、化学和药学等领域被广泛应用[5-11].化学位移与四极耦合常数(QCC)是反应物质结构信息的重要参数.化学位移能够反映核外电子,尤其是价电子对原子核的磁屏蔽效应;QCC主要反映了四极核所处的电场环境.对物质化学位移与QCC的表征可以反应出原子核所处的磁环境和电场环境,从而对物质结构进行明确的归属.其中17O NMR技术被广泛应用在无机、有机及生物体系的结构解析中[12-14].通过对δO、σO和QCC等NMR参数的正确解析可以获得体系的结构和功能信息.比如,Profeta等人[15]通过NMR技术对方晶石、石英等分子筛δO和QCC等NMR参数的表征检测,对分子筛结构进行了明确归属.Yamada等人[16]通过NMR技术对17O标记的三种氨基化合物的结构进行了研究,结果表明,三种氨基化合物中羰基17O QCC变化幅度较小,σO变化范围相对较大,该工作同时对电场梯度张量(EFG)和σO的取向进行了合理归属.
然而,实验的局限性对物质结构信息的正确归属带来一定困难.理论计算作为实验方法的补充手段,可以从原子分子水平上得到物质的NMR参数信息,结合实验上的结果,可以建立起其NMR实验数据与微观物质结构一一对应的关系[17-19].比如,Deng研究组[20]以理论计算为手段,通过计算得到的19F核的化学位移(δF)对实验中得到的不同金属氟化物活性位点进行了合理归属.Gervais等人[21]通过理论计算在对丙氨酸、酪氨酸及甘氨酸等结构研究中,计算得到的13C和15N核的屏蔽张量(σC、σN)和QCC等NMR参数与实验值非常吻合,并对氨基酸的NMR谱峰进行了合理归属.
选择合理的计算模型是保证获得正确的NMR参数的重要条件之一.有研究报导分子之间的氢键作用对计算中得到的NMR参数具有较大影响.例如,Uldry等人[22]发现,当不考虑分子间的氢键作用时,理论计算得到的麦芽糖和尿嘧啶中13C和15N核的化学位移(δC、δN)与实验结果误差分别有8.6和36.9;当考虑分子间氢键作用时,上述差异减小为2.1和17.9.Esrafili等人[23]通过理论计算证明了分子间氢键作用对壳聚糖中15N核的屏蔽张量具有较大的影响,在考虑分子间氢键作用之前,其δN与实验值相差8.92;在模型中包含氢键作用后,差值减小为7.39.由此可见,分子间的氢键作用对NMR参数具有显著的影响,只有在计算模型中充分考虑到了体系中的氢键作用,计算中得到的化学位移和QCC等NMR参数才比较准确,对物质的结构才能进行合理归属.
作为四极核,17O核由于核外电子对其周围局域环境[氢键、范德华(vdW)等作用力]较敏感,核外电子云易于极化,所以对实验值产生较大的误差.Wu等人[24]通过实验与密度泛函(DFT)计算相结合的方法对核酸碱基17O NMR参数进行了研究,然而小的团簇模型不能充分包含分子间的氢键作用,相对实验数据偏差将达到67,严重地制约了实验数据的合理归属.本文中,我们通过一系列包含不同氢键作用的核酸碱基模型研究了氢键对其中17O NMR参数的影响,并进一步揭示了产生这种影响的内在原因.
1 计算方法与模型 1.1 模型的选取本文所研究的四种核酸碱基结构见图 1.研究中所用的团簇模型以及周期性模型的初始结构均根据X射线晶体结构搭建[25-28].

|
图 1 ωB97XD/6-311++G(d, p)水平下优化得到的核酸碱基团簇模型Ⅴ结构.(a)胸腺嘧啶;(b)尿嘧啶;(c)胞嘧啶;(d)鸟嘌呤;距离数值单位为Å (0.1 nm). |
我们采用五种团簇模型来研究核酸碱基氢键作用对碱基17O NMR参数的影响.以尿嘧啶团簇模型为例:模型Ⅰ,即图 2(a),对应图 2(e)中1号分子;模型Ⅱ,即图 2(b),对应图 2(e)中1、2号分子;模型Ⅲ,即图 2(c),对应图 2(e)中1、3号分子;图 2(d)为团簇模型Ⅳ,对应图 2(e)中1、2、3号分子;图 2(e)为团簇模型Ⅴ.同样,对于胸腺嘧啶、胞嘧啶和鸟嘌呤,模型Ⅰ是单分子核酸碱基,分别对应图 1(a),1(c)和1(d)中1号分子;模型Ⅱ是双分子模型,胸腺嘧啶和胞嘧啶分别对应图 1(a)和1(c)中1、2号分子,鸟嘌呤对应图 1(d)中1、4号分子;模型Ⅲ,胸腺嘧啶和胞嘧啶分别包含图 1(a)和1(c)中1、3号分子,鸟嘌呤包含图 1(d)中1、2号分子和4号水分子;模型Ⅳ,胸腺嘧啶和胞嘧啶分别包含图 1(a)和1(c)中1、2和3号分子,鸟嘌呤包含图 1(d)中1、2、3号分子和4号水分子.为了考虑相邻其他分子之间的弱相互作用对中心分子的影响,我们同样构建了包含周围分子的模型Ⅴ(见图 1).

|
|
图 2 团簇模型ωB97XD/6-311++G(d, p)方法和周期性模型GGA/PBE方法下优化得到的尿嘧啶模型结构.(a)模型Ⅰ;(b)模型Ⅱ;(c)模型Ⅲ;(d)模型Ⅳ;(e)模型Ⅴ;(f)周期性模型;距离数值单位为Å (0.1 nm). |
传统小的团簇模型没有充分考虑到生物体系中的相互作用,会产生较大的误差.为了进一步研究片层之间弱的相互作用是否会对碱基中17O NMR参数产生影响,这里进一步研究了包含整体晶胞的周期性模型[见图 2(f)].
1.2 计算方法本文对五种团簇模型的计算都采用Gaussian 09软件包[29]完成.结构优化与NMR计算在ωB97XD泛函结合6-311++G(d,p)基组下进行.与传统的B3LYP相比,ωB97XD包含了描述长程色散作用的矫正函数,能够很好地描述分子之间的弱相互作用[30].由于大的模型结构使得在ωB97XD/6-311++G(d,p)水平下全优化难以完成,所以模型Ⅴ中将中心分子以及与中心分子有直接相互作用的周围原子进行优化,剩余的原子都被固定在原来的晶格位置.为了将体系中分子之间相互作用力充分考虑进来,这里进一步采用周期性模型进行了研究.几何结构优化及17O NMR参数计算都采用广义梯度函数(GGA)PBE[31]处理电子与电子之间的交换关联势[32, 33],并采用DFT-D算法引入色散校正项.结构优化在Material studio软件包中DMol3模块下完成.采用高精度布里渊区K点和高精度轨道截断能.当系统总能量变化稳定在2.72×10-4eV以内时.电子结构的自洽场(SCF)达到收敛标准停止计算,晶体内应力收敛标准是0.054 4 eV,最大位移收敛值是0.000 5 nm.在优化好的结构基础上,对NMR参数的计算由Material studio软件包中CASTEP模块完成,所有NMR参数计算都是基于GIPAW(gauge-including projector augmented wave)方法[34]完成.采用高精度布里渊区K点,平面波截断能设置为550 eV,能量变化稳定在1.0 × 10-6eV以内时,电子结构自洽场达到收敛标准停止计算,并在赝势on-the-fly上处理核与价电子的相互作用.
为了使屏蔽张量计算值(σcal)与实验值各向同性化学位移做直接比较,需要将σcal转化为各向同性化学位移值.以往对δO计算大多以水分子定标[35, 36],因水与核酸碱基结构差别较大,会较大的偏离实验值.本文对团簇模型和周期性模型的研究中以尿嘧啶来定标.尿嘧啶的17O核化学位移的实验值是275[24];在团簇模型和周期性模型计算得到的绝对屏蔽理论值是19和-10.因此,团簇模型和周期性模型计算对应于17O核化学位移定标分别对应于294和265.如下所示,我们得到了计算17O核屏蔽张量的公式:
团簇模型计算:
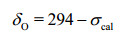 (1)
(1)
周期性模型计算:
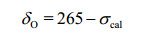 (2)
(2)
其中对于σcal,有σxx≤σyy≤σzz,这里以δ11≤δ22≤δ33表示.
同样,要把计算值电场梯度(EFG)张量部分转化为QCC:
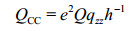 (3)
(3)
其中Q、e、qzz分别是17O核四极距、电子电荷、EFG张量z方向分量,这里Q我们采用标准值-2.558 fm2[37].
非对称因子(ηQ)计算如下:
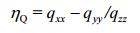 (4)
(4)
为了直观分析核酸碱基分子间存在的非共价相互作用,这里应用了由Yang等人[38]开发的非共价相互作用指示方法.在这种方法中,约化密度梯度(RDG)计算如下:
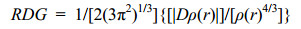 (5)
(5)
结合电子密度(ρ)用来判别分子间共价作用和非共价相互作用.其中非共价对应较低的电子密度和低的RDG值.λ2是电子密度Hessian矩阵第二大本征值,共价键作用时λ2 < 0,非共价作用时λ2 > 0.将指示函数sign[λ2(r)]ρ(r)用不同颜色投影到RDG等值面上即可显示出非共价相互作用的类型.氢键作用或强卤键作用时,sign[λ2(r)]ρ(r) < 0;弱的范德华力相互作用时,sign[λ2(r)]ρ(r)≈ 0;分子间强的排斥作用时,sign[λ2(r)]ρ(r) > 0.为了更明显地呈现核酸碱基分子间的非共价相互作用,这里消除了由RDG函数计算出的分子内相互作用.RDG和sign[λ2(r)]ρ(r)均由Multiwfn软件[39]计算完成.
我们同时进行了差分电荷密度分析,即成键前和成键后对应原子的电荷密度之差,由此可以定性分析成键过程中原子的电荷迁移信息.
2 结果与讨论根据文献报道,可视化的RDG等值面可以在实空间上定性地显示出分子之间弱的非键相互作用,这种方法已经被广泛用于主客体之间的弱相互作用分析[40, 41].图 3提供了胸腺嘧啶、尿嘧啶、胞嘧啶和鸟嘌呤模型Ⅴ中的RDG等值面图.由图 3(a)中的蓝色区域可知,邻近分子中的N-H基团与中心胸腺嘧啶分子之间的C=O基团之间存在着较强的氢键作用,同样的邻近分子中的C=O基团与中心胸腺嘧啶分子之间的N-H基团之间也存在着较强的氢键作用.除分子之间强氢键作用外,由图 3(a)中绿色区域可知,临近分子与中心胸腺嘧啶分子之间还存在弱的范德华作用,主要表现为C…H-C,C-H…O=C.对于图 3(b)尿嘧啶和图 3(c)胞嘧啶,与胸腺嘧啶一样,同样存在N-H基团与C=O基团之间的强氢键作用,以及C…H-C,C-H…O=C之间的范德华作用.另外,对于胞嘧啶,N…H-N之间同样表现为强的氢键作用.对于鸟嘌呤,临近分子对中心鸟嘌呤分子同样存在强的氢键作用以及弱的范德华作用,与图 3(a)、3(b)和3(c)不同的是,其中强的氢键作用还表现为中心鸟嘌呤O=C基团与4号水分子O-H基团之间的相互作用,弱的范德华作用还表现为中心分子C-H基团与5号水分子O-H基团之间的作用[见图 3 (d)].由此可见,对于核酸碱基来说,临近分子与中心分子之间普遍存在着氢键作用与范德华作用.根据文献[42, 43]报道,分子之间弱的相互作用对原子的NMR参数具有较大的影响.接下来,我们将采用六种不同程度上包含分子间弱相互作用的模型,来探索分子间氢键作用对四种核酸碱基17O NMR参数的影响,并揭示氢键作用影响NMR参数的机制.这将为理论计算中选择合适的计算模型提供参考.

|
| 图 3 分子间约化密度梯度(RDG=0.450 0 au)的等值面分析图,等值面颜色由sign[λ2(r)]ρ(r)和RGB数值大小定量绘制.(a)胸腺嘧啶;(b)尿嘧啶;(c)胞嘧啶;(d)鸟嘌呤 Fig. 3 Isosurface plots of reduced density gradient (RDG=0.4500 au) for nucleic acid bases.The isosurfaces of reduced density gradient are colored according to the values of the quantity sign[λ2(r)]ρ(r), and the RGB scale is indicated.(a) Thymine; (b) Uracil; (c) Cytosine; (d) Guanine |
表 1提供了胸腺嘧啶17O核在五种团簇模型以及周期性模型下的NMR参数.由表 1可见,从模型Ⅰ到模型Ⅳ,随着模型的逐渐增大,17O-2对应的δO逐渐降低,从模型Ⅰ时的272到模型Ⅳ时214,降低了58;值得注意的是,相对来说,17O-4的δO在模型的增加过程中的变化趋势并没有17O-2明显.由图 1(a)可知,随着模型的增大(从模型Ⅰ到模型Ⅳ),近邻的分子对中心胸腺嘧啶分子17O-2的氢键作用被逐渐考虑进来,而17O-4在模型的增加过程中[图 1(a)从模型Ⅰ到模型Ⅳ]并没有强氢键的加入.由此我们可以得出,分子间强的氢键作用对δO具有显著影响.同样地,分子间强的氢键作用对17O核的QCC也具有较大的影响.比如,当计算模型由单分子的Ⅰ增加到三分子的Ⅳ时,17O-2的QCC由8.383 MHz逐渐减小到7.366 MHz,降低了1.017 MHz;而17O-4 QCC从模型Ⅰ时9.629 MHz到模型Ⅳ时的9.864 MHz,仅仅变化了0.235 MHz(见表 1).值得注意的是,对于模型Ⅱ与Ⅲ 17O-2都只受到一个氢键作用的影响,但相比于模型Ⅱ来说,模型Ⅲ δO和QCC分别降低了δ12和0.1 MHz.通过对比我们发现,模型Ⅲ对应的氢键键长为0.178 4 nm,模型Ⅱ中的为0.183 5 nm.这种更强的氢键作用造成模型Ⅲ对应的δO和QCC变化更加明显.从上文分析可知分子间的氢键作用对计算中获得的δO和QCC具有一定的影响:氢键作用越强,δO和QCC越小[44].相似结论在之前包含氢键体系中也有过报道,例如,Ida等人[45]对尿嘧啶的结构进行研究时,发现17O NMR参数,随着模型的变化,体系中氢键作用逐渐增强,δO逐渐降低.
| Compound | modelb, c | δO | δ11 | δ22 | δ33 | QCC/MHz | ηQ | NPA(O)/|e| | NPA(C)/|e| |
| [17O-2]thymine | Ⅰ | 272 | 433 | 385 | 1 | 8.383 | 0.322 | -0.629 | 0.821 |
| Ⅱ | 244 | 378 | 344 | 11 | 7.965 | 0.598 | -0.683 | 0.835 | |
| Ⅲ | 232 | 361 | 327 | 9 | 7.865 | 0.681 | -0.702 | 0.839 | |
| Ⅳ | 214 | 315 | 297 | 30 | 7.366 | 0.971 | -0.749 | 0.850 | |
| Ⅴ | 216 | 320 | 299 | 28 | 7.504 | 0.928 | -0.746 | 0.850 | |
| PM | 206 | 317 | 282 | 18 | 7.299 | 0.877 | - | - | |
| Expt. | 200 | 290 | 270 | 20 | 6.650 | 1.000 | - | - | |
| [17O-4]thymine | Ⅰ | 372 | 684 | 470 | -37 | 9.629 | 0.026 | -0.608 | 0.668 |
| Ⅱ | 366 | 672 | 465 | -39 | 9.563 | 0.005 | -0.609 | 0.667 | |
| Ⅲ | 388 | 725 | 485 | -44 | 9.902 | 0.084 | -0.588 | 0.668 | |
| Ⅳ | 384 | 716 | 483 | -45 | 9.864 | 0.071 | -0.591 | 0.667 | |
| Ⅴ | 355 | 652 | 450 | -38 | 9.541 | 0.070 | -0.634 | 0.674 | |
| PM | 335 | 624 | 428 | -46 | 9.054 | 0.210 | - | - | |
| Expt. | 325 | 570 | 360 | 20 | 8.400 | 0.100 | - | - | |
| [17O-2]uracil | Ⅰ | 279 | 443 | 392 | 1 | 8.459 | 0.286 | -0.623 | 0.823 |
| Ⅱ | 284 | 452 | 403 | -3 | 8.525 | 0.249 | -0.615 | 0.822 | |
| Ⅲ | 286 | 461 | 400 | -2 | 8.627 | 0.243 | -0.615 | 0.825 | |
| Ⅳ | 290 | 465 | 406 | -1 | 8.627 | 0.227 | -0.608 | 0.824 | |
| Ⅴ | 257 | 403 | 361 | 8 | 8.326 | 0.484 | -0.600 | 0.851 | |
| PM | 258 | 419 | 356 | -2 | 8.161 | 0.511 | - | - | |
| Expt. | 245 | 400 | 330 | 10 | 7.610 | 0.500 | - | - | |
| [17O-4]uracil | Ⅰ | 378 | 676 | 478 | -21 | 9.672 | 0.028 | -0.595 | 0.659 |
| Ⅱ | 333 | 579 | 427 | -5 | 9.139 | 0.308 | -0.660 | 0.678 | |
| Ⅲ | 335 | 591 | 426 | -11 | 9.195 | 0.219 | -0.653 | 0.676 | |
| Ⅳ | 293 | 497 | 375 | 6 | 8.341 | 0.511 | -0.713 | 0.690 | |
| Ⅴ | 275 | 457 | 357 | 11 | 7.963 | 0.629 | -0.731 | 0.696 | |
| PM | 275 | 464 | 352 | 9 | 7.444 | 0.727 | - | - | |
| Expt. | 275 | 470 | 350 | 10 | 7.850 | 0.550 | - | - | |
| [17O-2]cytosine | Ⅰ | 309 | 485 | 425 | 17 | 8.961 | 0.169 | -0.633 | 0.793 |
| Ⅱ | 298 | 472 | 405 | 17 | 8.902 | 0.283 | -0.669 | 0.799 | |
| Ⅲ | 263 | 396 | 364 | 29 | 8.302 | 0.540 | -0.709 | 0.807 | |
| Ⅳ | 248 | 361 | 345 | 39 | 8.019 | 0.656 | -0.755 | 0.814 | |
| Ⅴ | 228 | 329 | 314 | 40 | 7.826 | 0.807 | -0.786 | 0.843 | |
| PM | 241 | 360 | 333 | 30 | 7.723 | 0.777 | - | - | |
| Expt. | 230 | 350 | 300 | 40 | 7.200 | 0.700 | - | - | |
| [17O-6]guanine | Ⅰ | 329 | 584 | 444 | -41 | 9.089 | 0.074 | -0.601 | 0.664 |
| Ⅱ | 293 | 526 | 399 | -45 | 8.664 | 0.308 | -0.653 | 0.671 | |
| Ⅲ | 277 | 480 | 375 | -23 | 8.623 | 0.491 | -0.696 | 0.673 | |
| Ⅳ | 252 | 445 | 344 | -32 | 8.390 | 0.574 | -0.734 | 0.682 | |
| Ⅴ | 251 | 439 | 341 | -27 | 8.381 | 0.583 | -0.742 | 0.695 | |
| PM | 227 | 400 | 312 | -30 | 7.419 | 0.857 | - | - | |
| Expt. | 230 | 395 | 285 | 10 | 7.100 | 0.800 | - | - | |
| a Calculated chemical shifts were converted from the computed shielding values using Eq.(1) for cluster models and Eq.(2) for periodic structure models. b Legends for experimental data: Expt.: experimental results taken from refs.[25~28]. c Cluster model Ⅰ、Ⅱ、Ⅲ、Ⅳ、Ⅴ and periodic structure (PM) model are included in Fig.1 and Fig.2. |
|||||||||
包含周围分子的模型Ⅴ不仅仅考虑到了分子间强的氢键作用还包含了弱的范德华作用.相对于简单的模型Ⅳ来说,在模型Ⅴ中17O-4受到临近分子C-H基团弱的范德华作用[见图 1(a)],导致δO和QCC相对于模型Ⅳ有了明显的降低,分别降低了δ29和0.323 MHz(见表 1).由此可见,不仅分子间强的氢键作用对δO与QCC具有影响,分子间弱的范德华作用同样会对计算得到的NMR参数造成影响.为了进一步考察片层之间的非键相互作用是否会影响计算得到的NMR参数,在本工作中同样考虑了包含整个晶体结构的周期性模型.计算得到的17O-2对应的δO与QCC分别为δ206、7.299 MHz,17O-4分别为δ335、9.054 MHz.相比模型Ⅴ,17O-2对应的δO和QCC分别下降了δ10、0.205 MHz,17O-4分别下降了δ20、0.487 MHz.而且与NMR实验上得到的数据(17O-2 δO,QCC:δ200、6.65 MHz;17O-4 δO,QCC:δ325、8.40 MHz)[24]非常接近.
2.1.2 对尿嘧啶δO和QCC的影响对于尿嘧啶,表 1同样提供了五种团簇模型以及周期性模型(见图 2)下17O的NMR数据.从表 1可见,单分子模型Ⅰ中17O-2对应的δO和QCC分别是δ 279、8.459 MHz,17O-4分别是δ378、9.672 MHz.显然,17O-4对应的δO大于17O-2,与胸腺嘧啶一样,这主要是由于17O-2连有两个共轭的类似吡咯的氮原子所造成的[见图 2(a)].与胸腺嘧啶相似,模型的增加对计算得到的尿嘧啶δO与QCC具有一定的影响.当计算模型由单分子的模型Ⅰ到三分子的模型Ⅳ时,17O-4 δO降低了85,17O-2 δO变化了11,这可以归因于模型的增加,分子间的非键相互作用逐渐包含进来所造成的.相较于17O-2,17O-4的δO变化的更加明显.与胸腺嘧啶相似,模型的增加对尿嘧啶分子中17O的QCC也具有一定的影响,这与δO的变化趋势一致.当模型由Ⅰ增加到Ⅳ时,17O-4降低了1.331 MHz,而17O-2只变化0.168 MHz.这主要是由于相对于17O-4来说,17O-2并没有受到分子间强的氢键作用的影响.虽然模型Ⅱ与模型Ⅲ中17O-4都只受到一个氢键作用的影响,但是相对于模型Ⅱ来说,模型Ⅲ中17O-4的δO与QCC分别增加了δ2和0.056 MHz,这主要是由于在这两个模型中17O-4受到的氢键作用力的不同.如图 2(b)、2(c)所示,模型Ⅱ中17O-4对应的氢键键长为0.180 7 nm,模型Ⅲ中氢键键长为0.181 6 nm,从模型Ⅱ到模型Ⅲ氢键作用强度变弱,对17O NMR参数的影响减弱,所以模型Ⅲ中δO和QCC相比模型Ⅱ均有增加.
如图 2(e)所示,对于模型Ⅴ,不仅仅包含了分子间强的氢键作用,还包含了弱的范德华作用.虽然在模型Ⅳ和模型Ⅴ中17O-4都受到两个氢键作用的影响,但相对于模型Ⅳ时的氢键长度0.180 3 nm(N-3-H…O-4=C-4)和0.186 7 nm(N-1-H…O-4=C-4),模型Ⅴ相应键长分别为0.180 3 nm和0.184 8 nm,表明在模型Ⅴ中氢键作用更强,造成17O-4 δO和QCC相对模型Ⅳ分别降低了δ18和0.378 MHz.值得注意的是,片层的模型中由于考虑了分子间弱的范德华相互作用(C-5-H…O-2=C-2=0.222 5 nm,C-6-H…O-2=C-2=0.217 9 nm),相较于模型Ⅳ,17O-2 δO和QCC分别降低了δ33和0.301 MHz,这与胸腺嘧啶中模型Ⅳ到模型Ⅴ时17O-4的δO和QCC的变化趋势相一致,进一步表明了分子间弱的氢键作用也会对17O NMR参数产生较大的影响.在考虑了整个晶体结构的周期性模型中,17O-2对应的δO和QCC分别是δ258、8.161 MHz,17O-4分别为δ275、7.444 MHz,相比模型Ⅴ,17O-2与17O-4对应的δO和QCC分别下降了δ-1、0.165 MHz和δ0、0.519 MHz,与17O-2和17O-4的实验值δ245、7.61 MHz和δ275、7.85 MHz[24]分别相差δ13、0.551 MHz和δ0、0.406 MHz.
2.1.3 对胞嘧啶δO和QCC的影响由图 1(c)所示,相比于胸腺嘧啶与尿嘧啶,胞嘧啶分子中存在一个羰基氧原子.由单分子模型Ⅰ计算得到的δO与QCC分别为δ309、8.961 MHz.据图 1(c)所示,当模型从Ⅱ增加到Ⅳ时,分子间的氢键作用被逐渐考虑进来.由前文胸腺嘧啶与尿嘧啶的结果可知,分子间强的氢键作用对与之相连的中心分子δO与QCC都具有较大的影响.比如,当模型增加到Ⅱ时,δO与QCC为δ298、8.902 MHz,相较于单分子的模型Ⅰ分别降低了δ11和0.059 MHz.模型Ⅱ和模型Ⅲ对应的氢键键长分别为0.232 0 nm和0.179 1 nm,显然后者氢键作用更强,对17O NMR参数的影响更大,所以从模型Ⅱ到模型Ⅲ δO从298降到263,QCC由8.902 MHz降到8.302 MHz,分别降低了35 MHz和0.6 MHz.
模型Ⅳ中17O-2涉及到两个氢键作用,氢键长度分别为0.203 0 nm [图 1(c) 1、2号分子]和0.192 0 nm [图 1(c) 1、3号分子],相比模型Ⅲ,氢键作用强度进一步增加,所以δO和QCC分别降低了δ15和0.283 MHz.片层的模型中由于加入了弱的范德华相互作用,对中心分子的影响增强,所以导致17O-2 δO和QCC进一步降低,相较于模型Ⅳ分别降低了δ20和0.193 MHz.对于考虑到整个分子间相互作用的周期性模型,17O-2 δO和QCC分别是δ241、7.723 MHz,相比模型Ⅴ,分别下降了δ-13、0.103 MHz,与实验值δ230、7.20 MHz[24]的差别分别是δ11和0.523 MHz.
2.1.4 对鸟嘌呤δO和QCC的影响与胞嘧啶相同,鸟嘌呤中同样只有一个羰基氧[见图 1(d)].单分子的模型Ⅰ中计算得到的17O-6的δO与QCC分别为δ329、9.089 MHz,当模型增大到Ⅳ时减小为δ252、8.390 MHz.不同于前文三种碱基,相较于模型Ⅱ,模型Ⅲ中心分子17O-6涉及两个氢键作用,导致模型Ⅲ相比模型Ⅱ,δO和QCC分别降低了δ16和0.041 MHz.模型Ⅲ中对应的两个氢键键长分别为0.191 9 nm(O-H…O-6=C-6)和0.211 2 nm(N-2-H…O-6=C-6),模型Ⅳ中其分别为0.177 7 nm和0.206 6 nm,所以模型Ⅳ中更强的氢键作用造成17O NMR参数从模型Ⅲ时的δ277、8.623 MHz到模型Ⅳ时δ252、8.390 MHz,分别降低了δ25和0.233 MHz.片层的模型中,虽然进一步增加了弱的范德华相互作用,然而这种作用对没有与其直接相互作用的17O-6影响并不明显,导致δO和QCC变化较小,分别降低了δ1、0.009 MHz.同样在周期性模型下,计算得到的17O核对应的δO和QCC分别是δ227和7.419 MHz,相比模型Ⅴ,分别下降了δ24、0.962 MHz,与实验值δ230、7.10 MHz[24]非常相近.
2.1.5 对4种核酸碱基对角值分量屏蔽张量的影响从表 1数据分析可知,随着模型的逐渐增大,屏蔽张量(δ11和δ22)与δO有相同的变化趋势,且变化幅度远大于δO.例如,对于鸟嘌呤,从模型Ⅰ到周期性模型,δ11和δ22分别降低了δ184、δ132,相应δO降低值只有δ102;又如,对于胞嘧啶,从模型Ⅰ到周期性模型,δ11下降了δ125,δ22下降了δ92,其δO只降低了δ68.同时还发现,分量δ33与δ11、δ22不同,随模型的增大没有明显的变化趋势.Yamada等人[16]在对酰胺化合物σO的研究中,他们认为分量δ33数值随氢键强度的加强而增大,这种差异很可能是由于B3LYP密度泛函方法没有考虑分子间弱相互作用而导致的.
从表 1结果还发现,ηQ与δO和QCC的变化趋势相反.例如,对于鸟嘌呤,模型Ⅰ时ηQ为0.074,模型Ⅴ时ηQ为0.583;随氢键作用的加强,δO和QCC不断降低,相应ηQ逐渐升高.所以从ηQ的角度也可以探究核酸碱基中氢键作用对17O NMR参数的影响[16].
2.2 模型选择及验证本文研究的一个重点是确定何种计算模型能够在计算中得到准确的17O NMR参数.根据文献报道,屏蔽张量的实验值与计算值的拟合关系可以定性地表达出计算值与实验值的吻合程度.比如:Yamada等人[16]在对氨基化合物中σO的研究中发现,相比单分子模型,二聚体化合物模型计算出的σO更接近实验值;另外,Hu等人[46]在对核酸碱基中σN的研究中同样发现,只有在包含更多的周围分子与中心分子有相互作用的模型中,屏蔽张量计算值才最吻合实验值.图 4提供了5种团簇模型和周期性模型下σcal与σexp关系.对于小的团簇模型Ⅰ、Ⅱ、Ⅲ和Ⅳ拟合斜率分别为1.399、1.314、1.313和1.224;截距分别为-25.165、-23.904、-29.747和-24.386,与实验数据拟合的差异大于模型Ⅴ时的1.118、-14.843.表明模型Ⅴ在计算中能得到与实验值比较吻合的结果.Ida等人[45]也表示在充分考虑了周围分子对中心分子作用的模型Ⅴ中,才能得到与σexp较吻合的结果.在周期性模型下计算得到的斜率与截距分别是1.115和-20.640,与模型Ⅴ的结果比较相似.然而,进一步通过对比模型Ⅴ和周期性模型σcal与σexp均方根(RMS)的差异发现,周期性模型的RMS(19.40)小于模型Ⅴ(25.57),这表明周期性模型下σcal与σexp结果更加吻合.为了进一步验证何种计算模型能够更加准确的得到实验的结果,我们同样得到计算中17O QCC与实验值的标准偏差.模型Ⅴ与实验值的标准差相差0.09 MHz;周期性模型下相差0.047 MHz.通过模型Ⅴ和周期性模型下δO和QCC NMR参数的对比表明,周期性模型能够得到与实验值更加吻合的结果.由图 5可见,随模型的增加分子间相互作用考虑越全面时,对应17O QCC越接近实验值.通过研究,我们发现虽然分子间的氢键作用是影响17O NMR参数的主要原因,但分子片层之间的弱的相互作用对碱基的17O NMR参数也具有一定的影响,所以只有综合考虑分子间强的氢键作用与弱的范德华相互作用,计算中才能得到与实验值更加吻合的结果,从而进一步对实验数据进行合理的归属[47].


|
| 图 5 不同计算模型理论预测的17O核的四极耦合常数 Fig. 5 Calculated 17O QCC with different structure models for [17O-2] thymine, [17O-4] uracil, [17O-2] cytosine and [17O-6] guanine |
从上文可以得到,随着分子间氢键作用的增强,δO逐渐降低.为了进一步揭示化学位移的变化与原子电荷的关系,这里我们将从NPA(natural population analysis)[48]电荷的角度来揭示氢键作用影响δO的内在原因.由于传统的Mulliken方法没有考虑到不同原子之间的差异并采取交叉项平分的处理方法,这样使得计算对基组有较大的依赖性,而在弥散基组下这些问题更加明显;而NPA方法将在原始基组下描述的波函数转化到正交极小基下描述,使得基函数和原子轨道之间有较为明确的对应关系,解决了计算方法对基组的依赖性问题,也避免了在处理交叉项时的困难[49].所以,这里我们采用NPA方法来研究氢键作用对原子电荷造成的影响.表 1提供了不同的计算模型中17O原子的NPA电荷变化情况.由表 1可知,随着计算模型的逐渐增大和氢键作用的加强,与氢键直接相连的17O原子的负电荷逐渐增加.比如,当计算模型由单分子的模型Ⅰ扩大到模型Ⅴ时,胸腺嘧啶17O-2负电荷由-0.629 |e|变化为-0.746 |e|;尿嘧啶17O-4从模型Ⅰ时的-0.595|e|变化到模型Ⅴ的-0.731|e|;胞嘧啶17O-2从模型Ⅰ时-0.633|e|变到模型Ⅴ时-0.786 |e|;鸟嘌呤17O-6由模型Ⅰ时-0.601|e|到模型Ⅴ时-0.742 |e|.众所周知,原子负电荷增加,会造成电子对核的屏蔽效应增强,δO减小.由此可见,氢键作用引起的17O原子负电荷密度逐渐增加是导致δO减小的内在原因.由图 6提供的17O原子NPA电荷与δO关系图可见,随着17O原子NPA负电荷的增加,核酸碱基δO逐渐降低.Gawinecki等人[50]在对亚硝胺分子间相互作用的研究中也发现17O原子NPA负电荷增加时,δO减小.通过进一步分析羰基13C原子上的NPA电荷,我们发现,17O原子上的负电荷之所以增加是由于随着氢键的增强,13C原子上的负电荷向17O原子上迁移所造成的.从图 7提供的尿嘧啶分子的差分电荷密度图上,我们也可以看到,当考虑到分子间的氢键作用时,羰基13C原子上的负电荷向17O原子上迁移(17O原子周围红色区域较明显,表明电子云密度增加,羰基13C原子周围的绿色区域表明电子云密度降低).Gawinecki等人[50]在对亚硝胺的核屏蔽效应与原子电荷相关性的研究中,也发现硝基中氧原子与氮原子NPA电荷存在相关.

|
| 图 6 17O NPA电荷[NPA(O)]与各向同性化学位移(δO)的关系 Fig. 6 The correlation between 17O negative charge density [NPA(O)] and isotropic chemical shifts (δO) |

|
| 图 7 尿嘧啶模型Ⅴ的差分电荷密度图 Fig. 7 Difference charge densities for cluster model Ⅴ of Uracil.The green represents where the electrons are coming from, and the red represents where the electrons are going |
17O NMR参数是表征核酸碱基微观结构的一种重要的手段.本工作通过高精度量子化学理论计算研究了分子间氢键以及范德华等弱相互作用对胸腺嘧啶、尿嘧啶、胞嘧啶和鸟嘌呤等四种核酸碱基σO和QCC等NMR参数的影响.研究结果表明,随着计算模型的逐渐增大,氢键作用逐渐增强,对应δO和QCC NMR参数不断降低.通过对周期性模型下δO和QCC等NMR参数的进一步研究表明,即使是分子间弱的范德华相互作用也会对17O核的NMR参数产生较大的影响.通过对模型Ⅴ和周期性模型下σcal与σexp的拟合关系发现,在综合考虑了多种拟合参数(斜率、截距、均方根和标准差)后发现,周期性模型下σcal与σexp最吻合,这说明只有在充分考虑了分子间强的氢键作用和弱的范德华相互作用的情况下,理论计算值才能更好的预测实验结果.通过对17O原子NPA电荷的研究表明,随着模型的增大,对应17O原子的负电荷密度也在不断增加,由此说明,氢键作用引起的17O原子的负电荷密度逐渐增加是导致δO减小的内在原因.
| [1] | Bedi D, Gillespie J W, Petrenko V A, et al. Targeted delivery of siRNA into breast cancer cells via phage fusion proteins[J]. Mol Pharm , 2013, 10 (2) : 551-559 DOI:10.1021/mp3006006 |
| [2] | Khandelwal G, Jayaram B. DNA-water interactions distinguish messenger RNA genes from transfer RNA genes[J]. J Am Chem Soc , 2012, 134 (21) : 8814-8816 DOI:10.1021/ja3020956 |
| [3] | Kierzek E, Pasternak A, Pasternak K, et al. Contributions of stacking, preorganization, and hydrogen bonding to the thermodynamic stability of duplexes between RNA and 2′-O-Methyl RNA with locked nucleic acids[J]. Biochemistry , 2009, 48 (20) : 4377-4387 DOI:10.1021/bi9002056 |
| [4] | Shaw N N, Arya D P. Recognition of the unique structure of DNA: RNA hybrids[J]. Biochimie , 2008, 90 (7) : 1026-1039 DOI:10.1016/j.biochi.2008.04.011 |
| [5] | Sardo M, Siegel R, Santos S M, et al. Combining multinuclear high-resolution solid-state MAS NMR and computational methods for resonance assignment of glutathione tripeptide[J]. J Phys Chem A , 2012, 116 (25) : 6711-6719 DOI:10.1021/jp302128r |
| [6] | Yao L, Grishaev A, Cornilescu G, et al. The impact of hydrogen bonding on amide 1H chemical shift anisotropy studied by cross-correlated relaxation and liquid crystal NMR spectroscopy[J]. J Am Chem Soc , 2010, 132 (31) : 10866-10875 DOI:10.1021/ja103629e |
| [7] | Nozad A G, Meftah S, Ghasemi M H, et al. Investigation of intermolecular hydrogen bond interactions in crystalline l-Cysteine by DFT calculations of the oxygen-17, nitrogen-14, and hydrogen-2 EFG tensors and AIM analysis[J]. Biophys Chem , 2009, 141 (1) : 49-58 DOI:10.1016/j.bpc.2008.12.013 |
| [8] | Morrissey J H, Tajkhorshid E, Sligar S G, et al. Tissue factor/factor Ⅶ a complex: role of the membrane surface[J]. Thromb Res , 2012, 129 (S2) : S8-S10 |
| [9] | Bechinger B, Resende J M, Aisenbrey C. The structural and topological analysis of membrane-associated polypeptides by oriented solid-state NMR spectroscopy: Established concepts and novel developments[J]. Biophys Chem , 2011, 153 (2, 3) : 115-125 |
| [10] | 戴晨晔, 刘买利, 李从刚. 低盐和高盐环境下α-Synuclein构象的19F NMN研究[J]. 波谱学杂志 , 2015, 32 (1) : 33-40 Dai Chen-ye, Liu Mai-li, Li Cong-gang. Salt content-dependent conformational changes of α-Synuclein studied by 19F NMR[J]. Chinese J Magn Reson , 2015, 32 (1) : 33-40 |
| [11] | 郁桂云, 彭路明. 固体核磁共振谱学研究层状双氢氧化物[J]. 波谱学杂志 , 2015, 32 (2) : 228-247 Yu Gui-yun, Peng Lu-ming. Solid-state NMR studies of layered double hydroxides: a review[J]. Chinese J Magn Reson , 2015, 32 (2) : 228-247 |
| [12] | Pike K J, Lemaitre V, Kukol A, et al. Solid-state 17O NMR of amino acids[J]. J Phys Chem B , 2004, 108 (26) : 9256-9263 DOI:10.1021/jp049958x |
| [13] | Lemaître V, de Planque M R R, Howes A P, et al. Solid-state 17O NMR as a probe for structural studies of proteins in biomembranes[J]. J Am Chem Soc , 2004, 126 (47) : 15320-15321 DOI:10.1021/ja0473283 |
| [14] | Wu G, Dong S. Two-dimensional 17O multiple quantum magic-angle spinning NMR of organic solids[J]. J Am Chem Soc , 2001, 123 (37) : 9119-9125 DOI:10.1021/ja0102181 |
| [15] | Profeta M, Mauri F, Pickard C J. Accurate first principles prediction of 17O NMR parameters in SiO2: assignment of the zeolite ferrierite spectrum[J]. J Am Chem Soc , 2003, 125 (2) : 541-548 DOI:10.1021/ja027124r |
| [16] | Yamada K, Dong S, Wu G. Solid-state 17O NMR investigation of the carbonyl oxygen electric-field-gradient tensor and chemical shielding tensor in amides[J]. J Am Chem Soc , 2000, 122 (47) : 11602-11609 DOI:10.1021/ja0008315 |
| [17] | Latosińska J N, Latosińska M, Tomczak M A, et al. Conformational stability and thermal pathways of relaxation in triclosan (Antibacterial/Excipient/Contaminant) in solid-state: Combined spectroscopic (1H NMR) and computational (Periodic DFT) study[J]. J Phys Chem A , 2015, 119 (20) : 4864-4874 DOI:10.1021/acs.jpca.5b02393 |
| [18] | 黄忆宁. 应用多核固体核磁共振光谱与量子化学计算的方法研究层状的过渡金属硫化物[J]. 波谱学杂志 , 2013, 30 (4) : 461-487 Sutrisno Andre, Huang Yi-ning. Multinuclear solid-state NMR and quantum chemical investigations of layered transition metal disulfides[J]. Chinese J Magn Reson , 2013, 30 (4) : 461-487 |
| [19] | ŁuczyńskaK, DrużbickiK, LyczkoK, 等. Experimental (X-ray, 13C CP/MAS NMR, IR, RS, INS, THz) and solid-state DFT study on (1:1) co-crystal of bromanilic acid and 2, 6-dimethylpyrazine[J]. J Phys Chem B , 2015, 119 (22) : 6852-6872 |
| [20] | Zheng A, Liu S B, Deng F. 19F Chemical shift of crystalline metal fluorides: Theoretical predictions based on periodic structure models[J]. J Phys Chem C , 2009, 113 (33) : 15018-15023 DOI:10.1021/jp904454t |
| [21] | Gervais C, Dupree R, Pike K J, et al. Combined first-principles computational and experimental multinuclear solid-state NMR investigation of amino acids[J]. J Phys Chem A , 2005, 109 (31) : 6960-6969 DOI:10.1021/jp0513925 |
| [22] | Uldry A C, Griffin J M, Yates J R, et al. Quantifying weak hydrogen bonding in iracil and 4-cyano-4'-ethynylbiphenyl: A combined computational and experimental investigation of NMR chemical shifts in the solid state[J]. J Am Chem Soc , 2008, 130 (3) : 945-954 DOI:10.1021/ja075892i |
| [23] | Esrafili M D, Elmi F, Hadipour N L. Density functional theory investigation of hydrogen bonding effects on the oxygen, nitrogen and hydrogen electric field gradient and chemical shielding tensors of anhydrous chitosan crystalline structure[J]. J Phys Chem A , 2007, 111 (5) : 963-970 DOI:10.1021/jp066761r |
| [24] | Wu G, Dong S, Ida R, et al. A Solid-state 17O nuclear magnetic resonance study of nucleic acid bases[J]. J Am Chem Soc , 2002, 124 (8) : 1768-1777 DOI:10.1021/ja011625f |
| [25] | Ozeki K, Sakabe N, Tanaka J. The crystal structure of thymine[J]. Acta Cryst, Section B , 1969, 25 (6) : 1038-1045 DOI:10.1107/S0567740869003505 |
| [26] | Stewart R F, Jensen L H. Redetermination of the crystal structure of uracil[J]. Acta Cryst , 1967, 23 (6) : 1102-1105 DOI:10.1107/S0365110X67004360 |
| [27] | Barker D L, Marsh R E. The crystal structure of cytosine[J]. Acta Cryst , 1964, 17 (12) : 1581-1587 DOI:10.1107/S0365110X64003899 |
| [28] | Thewalt U, Bugg C E, Marsh R E. The crystal structure of guanine monohydrate[J]. Acta Cryst, Section B , 1971, 27 (12) : 2358-2363 DOI:10.1107/S0567740871005880 |
| [29] | Frisch M J, Trucks G W, Schlegel H B, et al. Gaussian 09, B[M]. Wallingford: Gaussian Inc, 2009 |
| [30] | Chai J D, Head-Gordon M. Long-range corrected hybrid density functionals with damped atom-atom dispersion corrections[J]. Phys Chem Chem Phys , 2008, 10 (44) : 6615-6620 DOI:10.1039/b810189b |
| [31] | Perdew J P, Ernzerhof M, Burke K. Rationale for mixing exact exchange with density functional approximations[J]. J Chem Phys , 1996, 105 (22) : 9982-9985 DOI:10.1063/1.472933 |
| [32] | Perdew J P, Burke K, Ernzerhof M. Generalized gradient approximation made simple[J]. Phys Rev Lett , 1996, 77 (18) : 3865-3868 DOI:10.1103/PhysRevLett.77.3865 |
| [33] | Perdew J P, Burke K, Ernzerhof M. Perdew, burke, and Ernzerhof reply[J]. Phys Rev Lett , 1998, 80 (4) : 891-891 DOI:10.1103/PhysRevLett.80.891 |
| [34] | Pickard C J, Mauri F. All-electron magnetic response with pseudopotentials: NMR chemical shifts[J]. Phys Rev B , 2001, 63 (24) : 245101 DOI:10.1103/PhysRevB.63.245101 |
| [35] | Vaara J, Lounila J, Ruud K, et al. Rovibrational effects, temperature dependence, and isotope effects on the nuclear shielding tensors of water: A new 17O absolute shielding scale[J]. J Chem Phys , 1998, 109 (19) : 8388-8397 DOI:10.1063/1.477501 |
| [36] | Wasylishen R E, Mooibroek S, Macdonald J B. A more reliable oxygen-17 absolute chemical shielding scale[J]. J Chem Phys , 1984, 81 (3) : 1057-1059 DOI:10.1063/1.447799 |
| [37] | Pyykko P. Spectroscopic nuclear quadrupole moments[J]. Mol Phys , 2001, 99 (19) : 1617-1629 DOI:10.1080/00268970110069010 |
| [38] | Johnson E R, Keinan S, Mori-Sánchez P, et al. Revealing noncovalent interactions[J]. J Am Chem Soc , 2010, 132 (18) : 6498-6506 DOI:10.1021/ja100936w |
| [39] | Lu T, Chen F W. Multiwfn: A multifunctional wavefunction analyzer[J]. J Comp Chem , 2012, 33 (5) : 580-592 DOI:10.1002/jcc.v33.5 |
| [40] | Chu Y, Zheng A, Deng F. Slight channel difference influences the reaction pathway of methanol-to-olefins conversion over acidic H-ZSM-22 and H-ZSM-12 zeolites[J]. Catal Sci Technol , 2015, 5 (7) : 3507-3517 DOI:10.1039/C5CY00312A |
| [41] | Chu Y, Zheng A, Deng F. Strong or weak acid, which is more efficient for Beckmann rearrangement reaction over solid acid catalysts?[J]. Catal Sci Technol , 2015, 5 (7) : 3675-3681 DOI:10.1039/C5CY00619H |
| [42] | Danelius E, Andersson H, Brath U, et al. Solution NMR investigations of weak interactions using peptidomimetic templates[C]. San Francisco: 248th ACS National Meeting, 2014. |
| [43] | Bogle X, Vazquez R, Greenbaum S, et al. Understanding Li+-solvent interaction in nonaqueous carbonate electrolytes with 17O NMR[J]. J Phys Chem Lett , 2013, 4 (10) : 1664-1668 DOI:10.1021/jz400661k |
| [44] | Ropp J, Lawrence C, Farrar T C, et al. Rotational motion in liquid water is anisotropic: A nuclear magnetic resonance and molecular dynamics simulation study[J]. J Am Chem Soc , 2001, 123 (33) : 8047-8052 DOI:10.1021/ja010312h |
| [45] | Ida R, De Clerk M, Wu G. Influence of N-H…O and C-H…O Hydrogen bonds on the 17O NMR tensors in crystalline uracil: computational study[J]. J Phys Chem A , 2006, 110 (3) : 1065-1071 DOI:10.1021/jp0554947 |
| [46] | Hu J Z, Facelli J C, Alderman D W, et al. 15N Chemical shift tensors in nucleic acid bases[J]. J Am Chem Soc , 1998, 120 (38) : 9863-9869 DOI:10.1021/ja9816786 |
| [47] | Zheng A, Liu S B, Deng F. 13C shielding tensors of crystalline amino acids and peptides: Theoretical predictions based on periodic structure models[J]. J Comp Chem , 2009, 30 (2) : 222-235 DOI:10.1002/jcc.v30:2 |
| [48] | Reed A E, Weinstock R B, Weinhold F. Natural population analysis[J]. J Chem Phys , 1985, 83 (2) : 735-746 DOI:10.1063/1.449486 |
| [49] | 卢天, 陈飞武. 原子电荷计算方法的对比[J]. 物理化学学报 , 2012, 28 (1) : 1-18 Lu Tian, Chen Fei-wu. Comparison of computational methods for atomic charges[J]. Acta Phys-Chim Sin , 2012, 28 (1) : 1-18 |
| [50] | Gawinecki R, Kolehmainen E, Dobosz R, et al. Intramolecular interactions in nitroamines studied by 1H, 13C, 15N and 17O NMR spectral and quantum chemical methods[J]. J Iran Chem Soc , 2014, 11 (1) : 17-25 DOI:10.1007/s13738-013-0269-6 |