 2015, Vol. 32
2015, Vol. 32文章信息
- 丁丽红,刘小龙,王 强,刘文涛,朱诚身, 郑安民,邓 风
- DING Li-hong,LIU Xiao-long,WANG Qiang,LIU Wen-tao, ZHU Cheng-shen,ZHENG An-min,DENG Feng
- 多金属氧酸盐TBA3[VW5O19]及 多金属氧酸盐TBA3[VW5O19]及多金属氧酸盐TBA3[VW5O19]及的固体核磁共振研究的固体核磁共振研究
- Solid-State NMR Studies of TBA3[VW5O19] and TBA4[PVW11O40]
- 波谱学杂志, 2015, 32(3): 409-418
- Chinese Journal of Magnetic Resonance, 2015, 32(3): 409-418
- http://dx.doi.org/10.11938/cjmr20150302
-
文章历史
- 收稿日期:2014-06-27
- 收修改稿日期:2015-07-27






2. 波谱与原子分子物理国家重点实验室,武汉磁共振中心(中国科学院 武汉物理与数学研究所),湖北 武汉 430071
2. National Center for Magnetic Resonance in Wuhan, State Key Laboratory of Magnetic Resonance and Atomic and Molecular Physics (Wuhan Institute of Physics and Mathematics,Chinese Academy of Sciences), Wuhan 430071, China
多金属氧酸盐(Polyoxometalates,POMs)是由处于d0电子构型W(VI)、Mo(VI)、 V(V)、Nb(V)及Ta(V)等前过渡金属离子,以MO6为结构基元,通过共边或共角缩聚而成的多金属氧酸多核配合物.其中W和Mo是构成多金属氧酸盐的主要元素[1].目前已知可构成多酸的杂原子有70多种,而且组成多酸的杂原子通常以不同价态存在于多酸中,并且多酸阴离子是一类富氧阴离子,其中的氧原子易于参与配位,致使多酸化合物的种类和数目巨大.多金属氧酸盐可以是由同种含氧酸根离子缩合而形成的同多阴离子,其对应的酸称为同多酸;也可以是由不同种类的含氧酸根离子缩合而形成的杂多阴离子,其对应的酸称为杂多酸.多酸阴离子的基本结构有6种,它们是杂多酸阴离子的Keggin型、Dawson型、Anderson型、Waugh型、Silverton型和同多酸阴离子的Lindqvist型结构.
多金属氧酸盐,特别是具有Keggin构型的多金属氧酸盐,以其优异的物理化学性质,已经成为构造新型功能材料的重要结构基元.首先,多金属氧酸及其盐具有酸性、氧化-还原性或双功能性[2];其次,多金属氧酸盐阴离子具有不同的电荷、形状和尺寸,通过调变它和有机配体之间的相互作用,可以合成出具有多种骨架结构的新型催化材 料[3, 4];再次,多金属氧酸盐阴离子具有非常稳定的结构,可以通过改变分子组成和结构来调变其催化性能,以实现特定的催化要求[5, 6, 7].最后,多金属氧酸是一种环境友好的催化剂[8].因此,多金属氧酸盐作为新型固体酸催化剂[9, 10]在石油化工与精细化工中有着重要的应用,并且在医药、催化、功能材料、化学分析等领域也都有着广泛的应用.
同多酸的Lindqvist型结构和杂多酸的Keggin型结构是多金属氧酸盐最主要的,也是最重要的结构构型,特别是杂多酸的Keggin型结构在实际应用中有很大的应用价值,一直以来是理论与催化应用研究的重点.所以本文选择了四正丁胺钨钒酸盐(TBA3[VW5O19])及四正丁胺磷钨钒酸盐(TBA4[PVW11O40])晶体作为研究对象,因为它们分别具有Lindqvist构型和Keggin构型(图 1),通过固体核磁共振技术对它们的微观结构进行分析,进而理解结构对催化性能的影响.

|
| 图 1 钨钒酸 ([VW5O19]3-,Lindqvist)和磷钨钒酸([PVW11O40]4-,Keggin)阴离子的结构示意图 Fig. 1 The structure of [VW5O19]3- (Lindqvist) and [PVW11O40]4- (Keggin)anions |
Lindqvist结构[11]是由6个等同的MO6八面体围绕着中心的氧负离子形成的[M6O19]n-同多阴离子结构基元,整个阴离子的对称性为Oh.Lindqvist多酸化合物的最大结构特征是体系中含有相互垂直的含有4个金属原子的平面环,以及在金属与氧之间可能存在的(d-p)p共轭效应.这种特殊的六配位结构极大地影响了这一阴离子结构的电子性质以及几何结构[12, 13, 14, 15].Keggin阴离子[16]是由12个MO6八面体围绕着中心XO4四面体构成,呈现Td对称性.同时过渡金属取代会明显改变多酸化合物的化学反应活性,特别是氧化还原性质.对于多金属氧酸的进一步实验研究就是对多酸阴离子基本结构的修饰、多孔化、高核化、仿生化和纳米功能化,以得到具有应用潜力的功能材料.多酸的研究一直是材料化学和催化化学领域的热点.
钒取代的多酸阴离子具有很好的催化活性,特别是对烃类的氧化,它的活性主要受钒取代的数目和受阳离子环境影响的钒中心这两个因素影响[17, 18].本文所用的合成方法合成的是粉末多酸盐,相比于粉末XRD分析,固体核磁共振技术研究能够提供更加详细的结构信息.本文应用固体核磁技术分析——取代钒的两类典型结构中钒的局域结构和化学环境,以及有机阳离子对多酸阴离子结构的影响,特别是对钒的化学环境的影响,为研究多酸的催化活性和催化机理提供基本的结构信息.
1 实验部分 1.1 样品的制备 1.1.1 四正丁胺钨钒酸盐(TBA3[VW5O19])晶体的制备3.3 g Na2WO3·H2O和0.25 g NaVO3溶解于250 mL去离子水中,加热到80 ℃并保持5 min.然后缓慢加入20 mL 0.5 mol/L H2SO4到混合溶液中,并保持溶液温度在80 ℃下20 min.最后加入饱和的2.5 g TBABr[(n-C4H9)4NBr]溶液,缓慢冷却到室温.抽滤并洗涤黄色固体产物,然后用沸腾的乙腈重结晶.
1.1.2 四正丁胺磷钨钒酸盐(TBA4[PVW11O40])晶体的制备13.5 g H3PW12O40溶于12.5 mL去离子水中,加入固体Li2CO3到该溶液中并剧烈搅拌,调节pH值至4.9,再加入25 mL 0.2 mol/L NaVO3.然后逐滴加入6 mol/L的盐酸溶液调节混合溶液的pH值至2.0,加热溶液到60 ℃,并保持10 min,冷却到室温并再次逐滴加入6 mol/L的盐酸溶液调节混合溶液的pH值至2.0,并再次加热溶液到60 ℃.
最后加入4.03 g TBABr并维持溶液温度在60 ℃下10 min.缓慢冷却混合物到室温,抽滤并洗涤橙红色固体产物.
1.2 固体核磁共振实验
固体核磁共振(SSNMR)实验是在Bruker Avance-Ⅲ 500型核磁共振谱仪上进行.所用探头为4 mm HXY探头,13C、31P和51V的90°脉冲宽度分别为4.2、3.4和1.0 ms,测试温度为298 K,魔角旋转(MAS)的转速为10~12.5 kHz.51V、13C、31P化学位移定标的标样分别是饱和NaVO3溶液(dV = -578)、六甲苯(甲基dC = 17.55)和磷酸氢二铵(dP = 1).
2 结果与讨论作为四极核,51V的四极矩相对较小,而电子云密度较大,同时具有较高的旋磁比和天然丰度高(99.76%)的特点,因此具有很高的检测灵敏度,非常适合于核磁共振研究.51V NMR谱能够反映出核周围的电子环境,因而能够给出重要的结构信息.本文主要通过对51V MAS SSNMR谱的分析,并参考13C CP/MAS谱或31P MAS谱,研究杂多酸阴离子与TBA阳离子之间的相互作用及对它们周围环境的影响.杂多酸阴离子是一级结构,具有弱碱性,对反应物分子有特殊的配合能力,所以是影响杂多酸盐催化活性和选择性的重要因素.多酸阴离子与相应阳离子组成二级结构,阳离子的电荷、半径、电负性不同也会影响到催化性能.我们选择TBA作为多酸阴离子的反荷阳离子,是因为TBA只有一个正电荷,且半径大、电负性较低,它与多酸阴离子之间的相互作用较弱,所以51V NMR谱更能够体现出多酸阴离子结构对钒的影响.
2.1 四正丁胺钨钒酸盐(TBA3[VW5O19])晶体的固体核磁共振研究图 2(a)、(b)分别是TBA3[VW5O19]的XRD晶体粉末衍射谱和SEM图像.XRD晶体粉末衍射谱说明晶体相是纯相,但SEM图像显示仍然有无定型的颗粒在晶体的表面上.这些无定型的颗粒容易夹裹少量的TBAB r分子,而这些“杂质分子”很难被洗掉.尽管我们使用了超声和离心方法,但还是去除不了这些“杂质分子”.

|
| 图 2 (a) TBA3[VW5O19]的XRD谱图;(b) TBA3[VW5O19]的SEM谱图 Fig. 2 (a) XRD pattern of TBA3[VW5O19]; (b) SEM picture of TBA3[VW5O19] |
图 3所示的51V MAS SSNMR谱说明在晶体结构中钒只有一个晶格位点,即所有的钒都是化学和磁等价的;由于钒取代的多酸阴离子导致Lindqvist阴离子结构基元的对称性降低(由Oh高对称性降至C4v),这个结构对称性降低导致了钒具有较大的四极耦合常数(CQ)和化学位移的各向异性(Ωcsa).图 4是TBA阳离子的13C CP/MAS信号,碳谱也说明了TBA阳离子的化学和磁环境相同,且离氮原子越远的碳的信号越往高场移动;而其中的弱信号来自于样品的杂质(图中已用*标出).由于TBA结构作为阳离子相对于碱金属离子是相当大的,所以距离多酸表面存在不同的阴离子结合位点(如桥氧端氧)都是有相当距离的,这使得TBA结构对多酸表面存在不同的阴离子结合位点的“感受”是相同的.同时对比碳谱中“杂质”峰与多酸阴离子作用的TBA的半峰宽,“杂质”峰相对狭窄很多,我们认为这些“杂质”峰是不可能是源于与多酸表面的不同位点相结合的TBA阳离子.

|
| 图 3 TBA3[VW5O19]的51V MAS NMR谱,中心峰已用星号标出.其中CQ = 0.60 MHz,hQ = 0.65,dobsd = -418.8,Wcsa = 1 380,kcsa = 0.96,所有参数都通过拟合得到 Fig. 3 51V MAS NMR spectrum of TBA3[VW5O19],the center band is indicated with an asterisk. CQ = 0.60 MHz,hQ = 0.65,dobsd = -418.8,Wcsa = 1 380,kcsa = 0.96,all parameters were obtained from simulation |

|
| 图 4 TBA3[VW5O19]的13C CP/MAS NMR谱 Fig. 4 13C CP/MAS NMR spectrum of TBA3[VW5O19] |
图 5(a)、(b)所示的是TBA4[PVW11O40]的XRD晶体粉末衍射谱和SEM图像.XRD晶体粉末衍射谱和SEM图像说明得到的晶体相是纯的粉末多晶相.如果有多取代的多酸粉末晶体,它们具有很强的特征性XRD谱峰,但在XRD晶体粉末衍射谱并没有观测到这些信号.同时SEM图像显示粉末多晶相是比较均一的,所以初步判断得到的粉末晶体是一钒取代的多晶粉末.

|
| 图 5 (a) TBA4[PVW11O40]的XRD谱图;(b) TBA4[PVW11O40]的SEM谱图 Fig. 5 (a) XRD pattern of TBA4[PVW11O40]; (b) SEM picture of TBA4[PVW11O40] |
图 6所示的51V MAS SSNMR谱的两个信号说明在晶体结构中钒有化学等价而磁不等价的两个晶格位点(晶体学不等价)[19, 20],从而具有化学位移不同的两个信号(dV -548.5和-556.4).这是由于钒取代的多酸阴离子导致Keggin阴离子结构基元由Td的高对称性降低到Cs,结构对称性降低使得钒在与阳离子形成的晶体结构中具有化学等价而磁不等价的两个晶格位点,也导致了钒核较大的四极耦合常数(CQ)和化学位移的各向异性(Wcsa).化学等价是 因为晶体中阴阳离子是相同的,所以化学环境相同;而磁不等价是由于局部的对称性不同所造成的,化学位移不同并不是由于少量多钒取代晶体存在引起的.假设存在少量多钒取代晶体,它们的化学位移差会远大于8,且会有多个信号.

|
| 图 6 TBA4[PVW11O40]的51V MAS NMR谱,中心峰已用星号标出.其中CQ = 1.13和0.98 MHz,hQ = 0.61和0.52,dobsd = -548.5和-556.4,Wcsa = 1 680和1 620,kcsa = 0.96和0.95,所有参数都通过拟合得到 Fig. 6 51V MAS NMR spectrum of TBA4[PVW11O40],The center band is indicated with an asterisk. CQ=1.13 and 0.98,hQ = 0.61 and 0.52,dobsd = -548.5 and -556.4,Wcsa = 1 680 and 1 620,kcsa = 0.96 and 0.95,all parameters were obtained from simulation |
图 7中TBA阳离子的13C CP/MAS信号也说明了与磷钨钒酸阴离子对应的TBA阳离子也有环境不同的两个晶格位点;其中与氮靠近的亚甲基峰位差别明显,而较远的亚甲基和甲基峰位差别逐渐减少一直到消失.这是由于正丁基中碳的运动造成的,我们以与N的距离由近及远来标记C为C1,C2,C3,C4.对于磁不等价的两个碳信号,由于相互转化的运动会引起的两个不等价位的信号的化学位移和线型产生变化,而这一变化与运动频率有关.C1与N最近,受限最大,所以相互转化运动频率很低,接近于0.对于C2信号的强度与不等价的两个碳信号的比例关系有很大的不同,这一差别是由于运动频率较低(约38 Hz即0.1Δν1)造成的.而对于C3信号来说,由于不等价的两个碳信号转换频率接近于380 Hz造成两个谱峰重合的部分很大.对于C4信号,转换频率远大于380 Hz,所以只有一个信号.

|
| 图 7 TBA4[PVW11O40]的13C CP/MAS NMR谱 Fig. 7 13C CP/MAS NMR spectrum of TBA4[PVW11O40] |
同时,对于Lindqvist构型和Keggin构型的TBA阳离子中与氮相连的亚甲基的化学位移有d 1的不同,这说明了相同的阳离子与不同的多酸阴离子的静电相互作用是有差别的,而这一差别能够在碳谱中被观测到.
图 8是31P MAS NMR谱,磷处于Keggin阴离子结构基元中心,化学位移分别是15.1和15.6;这说明在晶体中磷也是存在于两个晶格位点上,且由峰面积比可知,在两个晶格位点的比例大致是3∶1,这是与51V谱和13C谱的结果相一致的.

|
| 图 8 TBA4[PVW11O40]的31P MAS NMR谱 Fig. 8 31P MAS NMR spectrum of TBA4[PVW11O40] |
由于四极相互作用的二阶诱导效应使得实验观测的化学位移与真实的各向同性化学位移有所不同,这一不同与核四极矩(e2qQ/h)、核的自旋量子数、核的Larmor共振频率、磁量子数(m)和化学位移的不对称因子(η)的关系如(1)式所示:
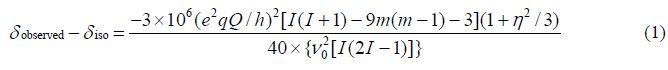
钒离子在Lindqvist和Keggin多酸结构中都是与氧负离子形成六配位的八面体结构,我们通过保持阳离子相同(TBA)的条件下,比较钒的化学位移在Lindqvist (-418.8)和Keggin (-548.5和-556.4)中的不同来进一步分析钒所受的Lindqvist和Keggin多酸结构的影响.由51V的化学位移对比可知,在Lindqvist结构中钒的电子云密度较低,这是由于钒与Lindqvist结构中心的氧之间存在的(d-p)p共轭效应,从而降低了钒的电子云密度,屏蔽效应减小;而在Keggin多酸结构则不存在这一相互作用.
3 结论本文论述了钒的化学位移与多酸阴离子结构的联系、阳离子在POM晶体中与多酸阴离子的相互作用及有机基团在晶体中的运动.由以上研究结果可知固体核磁共振能够有效用于钒取代POM晶体的结构解析,而这些微观结构信息将为多酸的催化活性和催化机理研究提供参考.多酸中的原子核特别是51V核的化学位移和四极耦合常数受环境(如阴离子配位结构和对应的阳离子结构)影响,具有较大变化范围,因而具有很高的检测灵敏度;这使得固体核磁共振能够成为研究钒取代的POM结构中的多酸阴离子构型简单且重要的表征手段.
| [1] | Wang En-bo(王恩波), Hu Chang-wen(胡长文), Xu Lin(许林). Introduction of Polyoxometalates Chemistry(多酸化学导论) [M]. Beijing(北京): Chemicel Industry Press(化学工业出版社出版), 1984. 4. |
| [2] | Yang C, Jin Q, Zhang H, et al. Tetra-(tetraalkylammonium)octamolybdate catalysts for selective oxidation of sulfides to sulfoxides with hydrogen peroxide[J]. Green Chem, 2009, 11: 1 401-1 405. |
| [3] | Johson B J S, Schroden R C, Zhuand C C, et al. Synthesis and characterization of 2D and 3D structures from organic derivatives of polyoxometalate clusters: Role of organic moiety, counterion and solvent[J]. Inorg Chem, 2001, 40(23): 5 972-5 978. |
| [4] | Chen X, Xu Z, Okuhara T. Liquid-phase esterification of acrylic acid with 1-butanol catalyzed by solid acid catalysts[J]. Appl Catal A, 1999, 180(1-2): 261-265. |
| [5] | Bareyt S, Piligkos S, Hasenknopf B, et al. Highly efficient peptide bond formation to functionalized wells-Dawson type polyoxotungstates[J]. Angew Chem Int Ed, 2003, 42(29): 3 404-3 406. |
| [6] | Carrier X, Caillerie J B, Lambert J F. The support as a chemical reagent in the preparation of Wox/y-Al2O3 catalysts formation and deposition of aluminotungstic heteropolyanions[J]. J Am Chem Soc, 1999, 121(14): 3 377-3 381. |
| [7] | Lu M, Wei Y G, Xu B B, et al. Hydrid molecular dumbbells: Bridging polyoxometalate clusters with an organic p-conjugated rod[J]. Angew Chem Int Ed, 2002, 41(9): 1 566-1 568. |
| [8] | Mizumo N, Misono M. Heterogeneous catalysis [J]. Chem Rev, 1998, 98(1): 199-205. |
| [9] | Xu Jun(徐君), Deng Feng(邓风). NMR studies on solid acids and crystallization of molecular sieves (固体酸催化剂及分子筛晶化过程的核磁共振研究)[J]. Chinese J Magn Reson(波谱学杂志), 2007, 24(3): 368-370. |
| [10] | Yu Zhi-wu(喻志武), Zheng An-min(郑安民), Deng Feng(邓风), et al. Acidity characterization of solid acid catalysts by solid-state NMR spectroscopy: A review on recent progresses(固体核磁共振研究固体酸催化剂酸性进展)[J]. Chinese J Magn Reson(波谱学杂志), 2010, 27(4): 485-515. |
| [11] | Lindqvist I. The structure of the hexanjobate ion in 7Na2O.6NB2O5.32H2O[J]. Arkiv Kemi, 1952, 5: 247 |
| [12] | Dmitrenko O, Huang W L, Polenova T E, et al. Effect of cations in infrared and computational analysis of vanadium-containing six-coordinate oxotungstates[J]. J Phys Chem B, 2003, 107(31): 7 747-7 752. |
| [13] | Derat E, Kumar D, Neumann R, et al. Catalysts for monooxygenations made from polyoxometalate: An iron(V)-oxo derivative of the lindqvist anion[J]. Inorg Chem, 2006, 45(21): 8 655-8 663. |
| [14] | Li J. Electronic structures, (d-p)p conjugation effects, and spectroscopic properties of polyoxometalates: M6O192- (M=Cr, Mo, W) [J]. J Clust Sci, 2002, 13(1): l37-l63. |
| [15] | Yang X, Waters T, Wang X B. Photoelectron spectroscopy of free polyoxoanions Mo6O192– in the gas phase[J]. J Phys Chem A, 2004, 108(46): 10 089-10 093. |
| [16] | Keggin J K. The structure and formula of 12-phosphotungstic acid[J]. Proc Roy Soc A, 1934, 144: 75-100. |
| [17] | Nomiya K, Nemoto Y, Hasegawa T, et al. Multicenter active sites of vanadium-substituted polyoxometalate catalysts on benzene hydroxylation with hydrogen peroxide and two reaction types with and without an induction period[J]. J Mol Catal A: Chemical, 2000, 152: 55-68. |
| [18] | Nomiya K, Hashino K, Nemoto Y, et al. Oxidation of toluene and nitrobenzene with 30% aqueous hydrogen peroxide catalyzed by vanadium(V)-substituted polyoxometalates[J]. J Mol Catal A, 2001, 176: 79-86. |
| [19] | Huang W, Todaro L, Yap G, et al. 51V magic angle spinning NMR spectroscopy of Keggin Anions [PVnW12-nO40](3+n)-: Effect of counteraction and vadadium substitution on fine structure constants[J]. J Am Chem Soc, 2004, 126: 11 564- 11 573. |
| [20] | Engelhardt G, Michel D. High-Resolution Solid-State NMR of Silicates and Zeolites[M]. Chichester: Wiley, 1987. |