 2022, Vol. 49
2022, Vol. 49文章信息
- 靶向药物联合应用对肝癌细胞SK-Hep-1增殖的影响及其机制
- Effects and Mechanisms of Combined Application of Molecular Targeted Drugs on Proliferation of Hepatocellular Carcinoma SK-Hep-1 Cells
- 肿瘤防治研究, 2022, 49(11): 1126-1133
- Cancer Research on Prevention and Treatment, 2022, 49(11): 1126-1133
- http://www.zlfzyj.com/CN/10.3971/j.issn.1000-8578.2022.22.0157
- 收稿日期: 2022-02-23
- 修回日期: 2022-08-02
引用本文 |



2. 046000 长治,长治医学院基础部生物化学与分子生物学教研室;
3. 030006 太原,山西白求恩医院普通外科;
4. 030013 太原,山西省肿瘤医院血液科
2. Department of Biochemistry and Molecular Biology, Changzhi Medical College, Changzhi 046000, China;
3. General Surgery Department, Shanxi Bethune Hospital, Taiyuan 030006, China;
4. Department of Hematology, Shanxi Cancer Hospital, Taiyuan 030013, China
对于单一驱动增殖的癌症,通过靶向相应的靶点,阻断增殖驱动因子,能够实现精准有效的治疗。例如,西妥昔单抗等药物通过靶向表皮生长因子受体(epidermal growth factor receptor,EGFR)有效治疗头颈部鳞状细胞癌[1-2]和非小细胞肺癌[3];伊马替尼通过靶向BCR-ABL酪氨酸激酶,能显著提高慢性髓性白血病患者的长期生存[4];曲妥珠单抗和帕妥珠单抗作用于HER2,可以显著降低早期HER2阳性乳腺癌患者脑转移的风险[5]。
然而,大多数实体瘤具有多驱动增殖的特点,单独阻断某一增殖驱动因子并不能抑制实体瘤的发展。前期研究发现,乳腺癌发病风险升高和12个基因的错义突变或蛋白截断变异密切相关[6];KRAS、NRAS、PI3KCA、BRAF等基因的变异累积能促进结肠癌肿瘤的发生发展[7-9];黑色素瘤中携带至少10个驱动因子突变[10]。
肝细胞癌(hepatocellular carcinoma, HCC)与多基因突变密切相关,这些突变引起相关信号通路的激活或抑制能影响肝细胞的异常增殖与分化[11-12]。研究表明,BRAF和PIK3CA基因突变在体细胞水平上促进肝癌的发生[13],肝癌细胞中RAS基因的低表达调节异常与肿瘤的异质性有关[14]。丝裂原活化蛋白激酶(mitogen activated protein kinase, MAPK)信号通路是BRAF和RAS基因发挥调控作用的基础,MAPK通路的下游信号系统为Ras/Raf/MEK/ERK,当机体因应激引起该信号通路激活时,这两种基因(BRAF和RAS两种突变在同一肿瘤细胞中同时表达的情况很少见,且即使在单细胞水平上同时表达,两者发挥作用也是相互独立的[15-16])就会发挥MAPK激酶激酶(MAPKKK)作用,进一步激活ERK,从而调控细胞的周期及凋亡[17]。PIK3CA突变可提高下游激酶PI3Ks的活性,激活PI3K/Akt/mTOR信号通路,减少细胞凋亡,并促进肿瘤浸润[18]。因此肝癌的发生是多驱动且多步骤的,进一步了解相关发病机制是挖掘潜在抗癌药物的基石。
本研究选用SK-Hep-1肝癌细胞株,从分子和细胞层面了解肝癌多驱动增殖的发病机制;通过建立更有效的数学模型,量化药物对细胞的作用;采用不同的组合,发掘有效的协同药物,为临床研究开发潜在的用药组合。
1 材料与方法 1.1 细胞株SK-Hep-1肝癌细胞购于中科院上海细胞库,细胞系STR(cell line authentication by STR profiling)鉴定正确,细胞用含10%胎牛血清的DMEM培养基于37℃、5%CO2培养箱培养。
1.2 药物本实验所用药物均为蛋白激酶抑制剂,见表 1。

DMEM培养基、胎牛血清均购于美国Hyclone公司;0.25%胰蛋白酶(含EDTA)、青链霉素混合液(100×)、MTT、DMSO、SDS、Tris、SDS-PAGE凝胶制备试剂盒、PVDF膜均购于北京索莱宝生物有限公司;Tween-20、RIPA蛋白裂解液、PMSF蛋白酶抑制剂、5×SDS-PAGE蛋白质上样缓冲液、BCA定量试剂盒均购于武汉博士德生物工程有限公司;一抗Src/p-Src、一抗Mek/p-Mek、一抗Erk/p-Erk、一抗PDK1/p-PDK1、一抗AKT/p-AKT、一抗BRAF/p-BRAF、一抗MET/p-MET均购于美国CST公司;一抗GAPDH购于北京中杉金桥生物有限公司;彩虹180广谱蛋白Marker(11~180 kD)购于北京Solarbio公司;辣根过氧化物酶标记羊抗兔二抗、辣根过氧化物酶标记羊抗鼠二抗均购于上海Santa Cruz公司;荧光显微镜购于日本Olympus公司;细胞培养箱、凝胶成像仪均购于美国Bio-Rad公司;酶标仪购于美国BioTek公司;蛋白电泳仪、垂直电泳槽、半干转膜仪均购于北京六一仪器厂。
1.4 MTT法测定细胞增殖能力将SK-Hep-1细胞按1×103个/孔接种至96孔板,24 h后加药处理,每种药物设16个浓度(20 μmol/L倍比稀释直至0.6 nmol/L)处理细胞,72 h后加入50 μl MTT溶液(5 mg/ml),避光孵育4 h。弃掉MTT,加入200 μl DMSO,使用BioTek酶标仪于490 nm和750 nm波长处检测每孔吸光度(OD)值。每个浓度设3个复孔,每个实验重复3次,使用SPSS22.0软件计算各种药物的IC50值。
1.5 细胞经药物处理后的动力学分析根据SK-Hep-1细胞对各种药物的IC50值,制作单靶点动力学分析曲线,定量了解SK-Hep-1细胞对药物的动态和复杂反应。单靶点动力学分析曲线遵守如下公式:I=[D]/([D]+IC50),I是相对抑制率,[D]是药物浓度。双相抑制曲线遵守如下公式:I=F1x[D]/([D]+Kd1)+F2 x[D]/([D]+Kd2)。I是细胞经药物处理后的增殖抑制率,F1是细胞中对药物敏感的部分,Kd是计算出来的最佳拟合参数,Kd1是敏感部分细胞的Kd,表示药物对靶细胞的亲和力,F2是细胞中对药物不敏感的部分,Kd2是不敏感部分细胞的Kd,表示药物对靶细胞的脱靶毒性。该双相公式要求F1+F2=1。拟合曲线为三次独立重复试验的平均结果,并计算拟合曲线的残差平方和(the sums of squared residues, SSR)。
1.6 Western blot实验SK-Hep-1细胞融合程度达到70%时,使用2.50 μmol/L crizotinib,2.50 μmol/L dasatinib,0.15 μmol/L HG6-64-1和12.00 μmol/L MK-2206处理SK-Hep-1细胞,2 h后提取细胞内总蛋白,运用BCA法在酶标仪中于562 nm波长处检测样品蛋白浓度,配置12%SDS-PAGE凝胶,上样、电泳,半干转至PVDF膜上,5%的脱脂奶粉封闭,TBST洗涤3次,孵育一抗(Src、p-Src、Mek、p-Mek、Erk、p-Erk、PDK1、p-PDK1、AKT、p-AKT、BRAF、p-BRAF、Met、p-Met、GAPDH)。4℃慢摇过夜,TBST洗涤3次,孵育二抗,室温慢摇孵育1 h,TBST洗涤3次,凝胶成像显影,Image J软件分析目的条带蛋白表达水平。
1.7 药物联合曲线绘制按1.3的方法对细胞进行药物联合处理后,计算IC50及IC70,并根据Chou-Talalay公式计算剂量减少指数(dose reduction index, DRI),评价药物组合与单药比较达到相同抑制水平减少药物浓度的倍数。
1.8 统计学方法使用SPSS22.0软件进行统计分析。所有实验均独立重复3次,数据以平均值±标准差显示。两个样本均数之间采用独立样本t检验,多个样本均数之间采用方差分析。P < 0.05为差异有统计学意义。
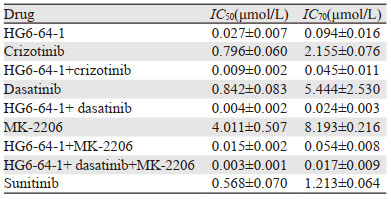
2 结果 2.1 SK-Hep-1细胞敏感药物确定实验结果显示:SK-Hep-1细胞对HG6-64-1敏感,IC50为0.027±0.007 μmol/L,但单药HG6-64-1对SK-Hep-1细胞的抑制率不能达到100%。dasatinib、crizotinib和sunitinib是能作用于不同信号通路靶点的蛋白激酶抑制剂,在本课题组先前的研究[19]中已经证实这三种药物能有效地抑制SK-Hep-1细胞的活力,且IC50分别为0.842±0.083、0.796±0.060和0.568±0.070 μmol/L,SK-Hep-1细胞对MK-2206不敏感,IC50为4.011±0.507 μmol/L,见表 2。这些研究表示SK-Hep-1细胞对多种药物敏感,提示其可能有多种独立驱动因子。
|
单靶点动力学分析结果显示:HG6-64-1和dasatinib对SK-Hep-1细胞的实际抑制情况与最佳拟合结果相比,SSR为0.187和0.432,拟合效果不佳。在低浓度药物范围,最佳拟合结果比实际抑制率高很多,而在高浓度药物范围,最佳拟合结果比实际抑制率又低很多。Crizotinib、sunitinib和MK-2206这三种药物处理的细胞中也发现了这种现象,但差异并不显著,SSR值分别为0.027、0.042、0.125,见图 1(左侧)。由此可见,单靶点动力学分析曲线不能准确反映SK-Hep-1细胞对这五种靶向药物的反应。

|
| A: HG6-64-1; B: dasatinib; C: crizotinib; D: sunitinib; E: MK-2206; The dose-response curves of SK-Hep-1 cells treated with different drugs using the single target kinetic formula (left) and biphasic inhibition formula (right). The blue closed points are the average values of three experiments, and the curve is the best fit result. Each inhibition curve is the average of three experiments. 图 1 SK-Hep-1细胞经蛋白激酶抑制剂抑制的动力学分析 Figure 1 Kinetic analysis of SK-HEP-1 cells inhibited by protein kinase inhibitors |
于是,我们猜测细胞对药物的反应是双相的。双相分析曲线显示:最佳拟合结果与实际抑制率拟合效果较好,SSR值分别为0.029、0.021、0.004、0.019、0.058,见图 1(右侧)。提示双相动力学曲线更能准确反映不同药物对SK-Hep-1细胞的抑制情况。
进一步分析双相变化,SK-Hep-1细胞经HG6-64-1处理后分为两部分:F1部分占(76.1±5.8)%,Kd1是10.8±6.7 nmol/L;F2部分占(23.9±5.8)%,Kd2是4.4±2.2 μmol/L。因F1是药物发挥抑制作用最重要的部分,几乎所有的抑制活动都在此处发生,可见76.8%的F1和10.4 nmol/L的Kd1比IC50=0.027 μmol/L更能准确反映药物和靶点之间的关系,见图 1A。SK-Hep-1细胞对其他药物的反应也能够类似地分解为这四个参数,见表 3。可见药物靶向特异性作用能阻断具有高亲和力的驱动因子使F1抑制,而药物的非特异性脱靶作用能导致F2抑制,这一结果与我们的猜测一致。

|
为验证HG6-64-1、dasatinib、MK-2206和crizotinib在SK-Hep-1细胞中阻断的驱动因素,我们检测了每种药物作用的关键信号蛋白水平。Western blot结果显示:crizotinib能强烈抑制Met磷酸化,也能较小程度抑制Akt磷酸化。HG6-64-1能完全抑制Akt磷酸化,部分抑制BRAF磷酸化和Mek磷酸化。dasatinib能完全抑制Y416位点和Y527位点Src磷酸化,也能较小程度抑制Mek和Erk磷酸化。MK-2206能完全抑制Akt磷酸化,也能够轻微抑制Mek和Erk磷酸化,见图 2。上述结果证明HG6-64-1、dasatinib、MK-2206和crizotinib通过抑制不同信号通路的关键成员阻断某一信号通路,对细胞增殖造成影响,并且也能交叉抑制相同通路,起到协同增强作用,也间接证明了SK-Hep-1细胞的多驱动增殖。

|
| 图 2 SK-Hep-1细胞经蛋白激酶抑制剂处理的信号通路分析 Figure 2 Signaling pathway analysis of SK-HEP-1 cells treated with protein kinase inhibitors |
通过双相分析得出,F1部分的数值越大,药物对细胞的抑制作用越好。但仅单药作用时,即使是SK-Hep-1细胞非常敏感的药物HG6-64-1也不能达到100%抑制。我们猜想将F1部分靶点不重合的抑制剂进行组合,可能会产生强协同效应。由表 3可知,10.8 nmol/L的HG6-64-1能够抑制约76%的SK-Hep-1细胞增殖,因此,我们试着寻找可以抑制剩余细胞增殖的药物。SK-Hep-1细胞中约30%对dasatinib敏感,约6%对crizotinib敏感,约7%对sunitinib敏感,约15%对MK-2206敏感,见表 3。将HG6-64-1分别与dasatinib、crizotinib和sunitinib联用,结果显示,从剂量-效应曲线可见:在低浓度区间(6.104×10-4 μmol/L~6.25×10-1 μmol/L),HG6-64-1与dasatinib的协同作用要优于HG6-64-1与crizotinib和HG6-64-1与MK-2206,HG6-64-1与sunitinib没有协同作用(数据未显示);在高浓度(1.25 μmol/L~20 μmol/L)区间,HG6-64-1与crizotinib的协同作用更优,见图 3。从剂量减量曲线可见:细胞存活率为10%时,DRI约为2;细胞存活率为70%时,DRI升高至6左右,可知细胞存活率为70%时,HG6-64-1和crizotinib两药联合的效果约为单药处理的6倍,见图 3A。细胞存活率为20%时,DRI约为5;细胞存活率为60%时,DRI升高至14左右,可知细胞存活率为60%时,HG6-64-1和dasatinib两药联合的效果约为单药处理的14倍,见图 3B。因此,我们可以证实HG6-64-1和dasatinib两药联合对SK-Hep-1细胞增殖的抑制作用更佳,在0.6 μmol/L时抑制率能达到30%~40%。

|
| The responses of SK-Hep-1 cells to different drug combinations and single drugs were compared. A: HG6-64-1 in combination with crizotinib; B: HG6-64-1 in combination with dasatinib; C: combined use of HG6-64-1 and MK-2206; D: HG6-64-1 combined with dasatinib and MK-2206. The experimental results are the average value of three experiments. The dose reduction index (DRI) chart is used to show the inhibition level. 图 3 靶向不同蛋白激酶药物对SK-Hep-1细胞增殖的的协同抑制作用 Figure 3 Synergistic inhibitory effect of different protein kinase drugs on SK-Hep-1 cell proliferation |
进一步分析HG6-64-1与dasatinib联用的结果,我们发现:在低浓度区间,dasatinib能使SK-Hep-1细胞中对HG6-64-1敏感的部分变得更敏感;在高浓度区间对HG6-64-1不敏感的部分对dasatinib也不敏感,见图 3B。为了使高浓度区间药物对细胞的抑制作用更显著,我们需选用一种可以使HG6-64-1和dasatinib联用也不敏感的部分变得敏感的药物。而在高浓度区间,crizotinib显示与HG6-64-1药物联合的协同作用更好,于是我们将crizotinib与dasatinib和HG6-64-1联合处理SK-Hep-1细胞,但结果显示协同作用不显著(数据未显示)。因此,我们从不同药物作用靶点出发,根据图 2可知:crizotinib能够完全抑制Met磷酸化;HG6-64-1能够部分抑制BRAF磷酸化,完全抑制Akt磷酸化;dasatinib是一种Src抑制剂。于是我们筛选靶点是Akt但F1高于10%的药物。MK-2206是Akt的特异性抑制剂,F1约为15%,将HG6-64-1与dasatinib和MK-2206联用,观察能否降低高浓度区域不敏感的细胞比例。结果显示:MK-2206显著降低了高浓度区间对HG6-64-1不敏感的细胞比例,见图 3C。HG6-64-1、dasatinib和MK-2206单药的IC50分别为0.027±0.007、0.842±0.083和4.011±0.507 μmol/L,IC70分别为0.094±0.016、5.444±2.530和8.193±0.216 μmol/L,而三药联合的IC50仅为0.003±0.001 μmol/L,IC70仅为0.017±0.009 μmol/L,见图 3D、表 2。以上结果表明,HG6-64-1、dasatinib和MK-2206三药联用能够有效抑制SK-Hep-1细胞增殖。
3 讨论肝癌SK-Hep-1细胞株本身具有BRAFV600E突变[20-21],BRAF是MAPK(Ras-Raf-MEK-ERK)通路的成员,并通过该通路介导细胞对生长信号的反应,特别是激活BRAF能参与细胞周期进程的持续诱导[13]。HG6-64-1是有效选择性的BRAF抑制剂[22],能显著抑制BRAF磷酸化激活。SK-Hep-1细胞对HG6-64-1非常敏感,IC50值仅为0.027±0.007 μmol/L。在高侵袭性癌细胞系中,受体酪氨酸激酶能够促进肿瘤细胞的迁移和侵袭,且在肝癌SK-Hep-1细胞中也已被证实通过化学抑制或mRNA沉默受体酪氨酸激酶家族中的DDR1(discoidin domain receptorⅠ)能够降低AKT和ERK磷酸化,从而减少癌细胞的增殖和迁移[23]。本课题组已从一组酪氨酸激酶抑制剂中筛选出三种对SK-Hep-1细胞敏感的抑制剂,分别为dasatinib、crizotinib和sunitinib[19]。因此,我们推测SK-Hep-1肝癌细胞有多种独立驱动因子参与细胞增殖。
一般使用IC50值反映细胞对药物的敏感度,并根据IC50值绘制单靶点抑制曲线。但由图 1可知,单靶点抑制曲线与每种药物的实际抑制作用之间存在偏差,说明单靶点抑制曲线在本研究中并不能准确描述药物作用情况。癌细胞一般由多个致癌因子独立驱动,使用靶向某个靶点的激酶抑制剂只能部分抑制细胞增殖,使用某浓度的激酶抑制剂也可能同时影响成百上千个蛋白激酶。受影响的某些激酶可能与细胞增殖不直接相关,但抑制这些激酶可能会导致细胞毒性。因此可见,多驱动增殖细胞对某一激酶抑制剂的反应是两种作用的混合:当药物抑制细胞敏感部分时,药物对细胞存在靶点特异性;当药物作用于细胞不敏感部分时,药物对细胞存在脱靶毒性。靶点特异性抑制是靶向治疗的基础,但脱靶毒性可能会限制靶向治疗的有效性。综上,准确区分靶点特异性抑制和脱靶毒性有助于更好地开发针对多驱动增殖细胞的联合靶向治疗策略。
本文选用双相数学模型,定量描述多驱动增殖SK-Hep-1肝癌细胞对靶向药物治疗的反应。双相动力学模型中F1、F2分别表示癌细胞中对某一激酶机制剂的敏感和抗性部分;Kd1表示药物对靶细胞的亲和力;Kd2表示药物对靶细胞的脱靶毒性。如果一个靶细胞是单驱动增殖的,药物反应就变成了一种极端情况,F1变成了100%或接近100%,F2阶段消失。在这种情况下增大F1的占比,药物对细胞的抑制作用越明显,脱靶毒性越小,治疗效果越好。一种癌细胞有多个独立驱动因素时,如果有一种完全可以与驱动因素单独作用的理想靶向药物,该药也只能部分抑制细胞增殖。当一种以上具有明显F1效应的药物对癌细胞有抑制作用时,将这些药物联合使用能更有效地阻断细胞活力,实现药物剂量显著降低。因此,双相分析不仅能有效评估多驱动增殖癌细胞对药物的反应,也能为靶向治疗提供新的框架。
大多数癌症依赖多驱动因素实现增殖,需要合理的药物组合进行靶向治疗。常规的药物组合离不开经验筛选和不断探索。本研究将HG6-64-1、dasatinib和MK-2206三药联用,发现三药联合的IC50和IC70与单药处理相比显著降低,说明这三药组合能够有效抑制SK-Hep-1细胞增殖。通过机制研究发现,这三种药物的作用靶点既有重合,也有不同,能够显著抑制Akt磷酸化和Src磷酸化,部分抑制BRAF磷酸化和Mek磷酸化。这些靶点与Ras/Raf/Mek/Erk和PI3K/Akt/mTOR信号通路有关,说明SK-Hep-1细胞增殖与这两种通路密切相关,与之前的报道一致[24]。我们先前的研究也证实dasatinib对SK-Hep-1细胞最可能的靶点是Src家族激酶[18],因此本次的药物组合结果有效。相关研究也证实,HGF/Met/JNK信号通路在肝硬化向肝癌发展的过程中具有一定作用[25]。Crizotinib是有效的c-Met抑制剂[26],单药也能够抑制SK-Hep-1细胞增殖(IC50=0.796±0.060 μmol/L, IC70=2.155±0.076 μmol/L),但抑制效果不明显,说明Met并不是SK-Hep-1肝癌细胞增殖的主要靶点。Western blot结果表明,crizotinib与dasatinib和HG6-64-1联合使用,多数作用靶点重叠,crizotinib能部分抑制Akt磷酸化,而HG6-64-1能完全抑制Akt磷酸化,说明这三种药物的结合并不互补,协同作用不显著。本实验中HG6-64-1、dasatinib和MK-2206这三种药物联用仅在细胞和分子层面表明对SK-Hep-1细胞增殖的有效抑制,今后有望进一步通过在体实验探讨药物联用对肝癌的有效性。
综上所述,对于多驱动肝癌细胞SK-Hep-1而言,双相驱动分析能更好地反映药物对细胞的抑制作用。HG6-64-1、dasatinib和MK-2206三药联合应用能有效地抑制SK-Hep-1细胞增殖,可能是一种潜在治疗肝癌的药物组合。
作者贡献:
朱晓霞:实验操作、数据分析及文章撰写
贾瑜琦:实验操作及文章校对
刘畅:实验设计及写作指导
弓韬:文献检索、筛选及基金支持
李高鹏:写作指导及基金支持
张宏伟:实验结果解读
于保锋:指导实验及论文审阅
| [1] |
Alvarado D, Ligon GF, Lillquist JS, et al. ErbB activation signatures as potential biomarkers for anti-ErbB3 treatment in HNSCC[J]. PLoS One, 2017, 12(7): e0181356. DOI:10.1371/journal.pone.0181356 |
| [2] |
Swick AD, Prabakaran PJ, Miller MC, et al. Cotargeting mTORC and EGFR Signaling as a Therapeutic Strategy in HNSCC[J]. Mol Cancer Ther, 2017, 16(7): 1257-1268. DOI:10.1158/1535-7163.MCT-17-0115 |
| [3] |
Karlsen EA, Kahler S, Tefay J, et al. Epidermal Growth Factor Receptor Expression and Resistance Patterns to Targeted Therapy in Non-Small Cell Lung Cancer: A Review[J]. Cells, 2021, 10(5): 1206. DOI:10.3390/cells10051206 |
| [4] |
Druker BJ. Translation of the Philadelphia chromosome into therapy for CML[J]. Blood, 2008, 112(13): 4808-4817. DOI:10.1182/blood-2008-07-077958 |
| [5] |
Duchnowska R, Loibl S, Jassem J. Tyrosine kinase inhibitors for brain metastases in HER2-positive breast cancer[J]. Cancer Treat Rev, 2018, 67: 71-77. DOI:10.1016/j.ctrv.2018.05.004 |
| [6] |
Breast Cancer Association Consortium, Dorling L, Carvalho S, et al. Breast Cancer Risk Genes-Association Analysis in More than 113, 000 Women[J]. N Engl J Med, 2021, 384(5): 428-439. DOI:10.1056/NEJMoa1913948 |
| [7] |
Fransén K, Klintenäs M, Osterström A, et al. Mutation analysis of the BRAF, ARAF and RAF-1 genes in human colorectal adenocarcinomas[J]. Carcinogenesis, 2004, 25(4): 527-533. |
| [8] |
Markowitz SD, Bertagnolli MM. Molecular origins of cancer: Molecular basis of colorectal cancer[J]. N Engl J Med, 2009, 361(25): 2449-2460. DOI:10.1056/NEJMra0804588 |
| [9] |
Janakiraman M, Vakiani E, Zeng Z, et al. Genomic and biological characterization of exon 4 KRAS mutations in human cancer[J]. Cancer Res, 2010, 70(14): 5901-5911. DOI:10.1158/0008-5472.CAN-10-0192 |
| [10] |
Tolcher AW, Peng W, Calvo E. Rational Approaches for Combination Therapy Strategies Targeting the MAP Kinase Pathway in Solid Tumors[J]. Mol Cancer Ther, 2018, 17(1): 3-16. DOI:10.1158/1535-7163.MCT-17-0349 |
| [11] |
邬宇美, 骆子义, 黄华. 原发性肝癌中KRAS及BRAF基因突变检测及临床意义分析[J]. 新发传染病电子杂志, 2018, 3(1): 37-40. [Wu YM, Luo ZY, Huang H. Detection of KRAS and BRAF gene mutations in primary hepatocellular carcinoma and clinical significance[J]. Xin Fa Chuan Ran Bing Dian Zi Za Zhi, 2018, 3(1): 37-40.] |
| [12] |
曹广文. "癌症进化发育学"理论进展及其在肝细胞癌靶向/免疫治疗中的作用[J]. 肿瘤防治研究, 2022, 49(8): 747-755. [Cao GW. Theoretical Update of Cancer Evo-Dev and Its Role in Targeted Immunotherapy for Hepatocellular Carcinoma[J]. Zhong Liu Fang Zhi Yan Jiu, 2022, 49(8): 747-755.] |
| [13] |
Colombino M, Sperlongano P, Izzo F, et al. BRAF and PIK3CA genes are somatically mutated in hepatocellular carcinoma among patients from South Italy[J]. Cell Death Dis, 2012, 3(1): e259. DOI:10.1038/cddis.2011.136 |
| [14] |
Bose S, Sakhuja P, Bezawada L, et al. Hepatocellular carcinoma with persistent hepatitis B virus infection shows unusual downregulation of Ras expression and differential response to Ras mediated signaling[J]. J Gastroenterol Hepatol, 2011, 26(1): 135-144. DOI:10.1111/j.1440-1746.2010.06305.x |
| [15] |
Sensi M, Nicolini G, Petti C, et al. Mutually exclusive NRASQ61R and BRAFV600E mutations at the single-cell level in the same human melanoma[J]. Oncogene, 2006, 25(24): 3357-3364. DOI:10.1038/sj.onc.1209379 |
| [16] |
Gray-Schopfer V, Wellbrock C, Marais R. Melanoma biology and new targeted therapy[J]. Nature, 2007, 445(7130): 851-857. DOI:10.1038/nature05661 |
| [17] |
Allegra CJ, Rumble RB, Schilsky RL. Extended RAS Gene Mutation Testing in Metastatic Colorectal Carcinoma to Predict Response to Anti-Epidermal Growth Factor Receptor Monoclonal Antibody Therapy: American Society of Clinical Oncology Provisional Clinical Opinion Update 2015 Summary[J]. J Oncol Pract, 2016, 12(2): 180-181. DOI:10.1200/JOP.2015.007898 |
| [18] |
Garcia-Echeverria C, Sellers WR. Drug discovery approaches targeting the PI3K/Akt pathway in cancer[J]. Oncogene, 2008, 27(41): 5511-5526. DOI:10.1038/onc.2008.246 |
| [19] |
Liu C, Zhu X, Jia Y, et al. Dasatinib inhibits proliferation of liver cancer cells, but activation of Akt/mTOR compromises dasatinib as a cancer drug[J]. Acta Biochim Biophys Sin (Shanghai), 2021, 53(7): 823-836. DOI:10.1093/abbs/gmab061 |
| [20] |
Chavda J, Bhatt H. Systemic review on B-RafV600E mutation as potential therapeutic target for the treatment of cancer[J]. Eur J Med Chem, 2020, 206: 112675. DOI:10.1016/j.ejmech.2020.112675 |
| [21] |
Davies H, Bignell GG, Cox C, et al. Mutations of the BRAF gene in human cancer[J]. Nature, 2002, 417(6892): 949-954. DOI:10.1038/nature00766 |
| [22] |
GRAY NS. Type Ⅱ Raf kinase inhibitors: US, WO2011090738A2[P]. 2011-07-28.
|
| [23] |
Romayor I, Badiola I, Olaso E. Inhibition of DDR1 reduces invasive features of human A375 melanoma, HT29 colon carcinoma and SK-HEP hepatoma cells[J]. Cell Adh Migr, 2020, 14(1): 69-81. DOI:10.1080/19336918.2020.1733892 |
| [24] |
Gao JJ, Shi ZY, Xia JF, et al. Sorafenib-based combined molecule targeting in treatment of hepatocellular carcinoma[J]. World J Gastroenterol, 2015, 21(42): 12059-12070. DOI:10.3748/wjg.v21.i42.12059 |
| [25] |
彭全斌, 朱书渊, 汪望月. HGF/Met/JNK信号通路介导的细胞自噬在肝硬化癌变进程中的作用[J]. 世界华人消化杂志, 2021, 29(20): 1158-1166. [Peng QB, Zhu SY, Wang WY. Autophagy mediated by the HGF/Met/JNK signaling pathway is involved carcinogenesis of liver cirrhosis[J]. Shi Jie Hua Ren Xiao Hua Za Zhi, 2021, 29(20): 1158-1166.] |
| [26] |
Cui JJ, Tran-Dubé M, Shen H, et al. Structure based drug design of crizotinib (PF-02341066), a potent and selective dual inhibitor of mesenchymal-epithelial transition factor (c-MET) kinase and anaplastic lymphoma kinase (ALK)[J]. J Med Chem, 2011, 54(18): 6342-6363. DOI:10.1021/jm2007613 |