 2021, Vol. 48
2021, Vol. 48文章信息
- 胰腺炎——癌转化的表观遗传学研究进展
- Research Progress in Epigenetics of Pancreatitis-cancer Transformation
- 肿瘤防治研究, 2021, 48(3): 219-223
- Cancer Research on Prevention and Treatment, 2021, 48(3): 219-223
- http://www.zlfzyj.com/CN/10.3971/j.issn.1000-8578.2021.20.0977
- 收稿日期: 2020-08-18
- 修回日期: 2020-12-18
引用本文 |



胰腺癌是恶性程度最高的肿瘤之一,五年生存率不足10%,诊断和治疗非常困难。全球肿瘤统计数据结果显示[1],2018年新诊断为胰腺癌的病例有458 918例,因胰腺癌死亡病例为432 242例,发病率和死亡率在全球范围内迅速上升,据美国癌症统计协会预测2030年胰腺癌将成为全球第二大癌症死因。近年来我国胰腺癌的疾病负担也逐渐加重,其5年生存率仅为7.2%,且逐年恶化[2]。胰腺癌的主要治疗方法为手术治疗,但由于胰腺癌早期指征的非特异性,缺乏早期诊断方法,且在早期可能已经发生微观转移[3],这导致胰腺癌确诊时已有超过80%的病例不可切除[4],同时也限制了其他局部治疗如放疗的有效性。因此胰腺癌传统的生物标志物或早期诊断方法已不适用,我们需要对它进行更早发现和干预。
慢性炎性反应是重要的癌症危险因素,全世界约五分之一的癌症发病与慢性炎性反应有关[5]。慢性胰腺炎是最公认的胰腺癌危险因素之一,有5%的慢性胰腺炎经过20年的发展可能进展为胰腺癌[6]。长期的炎性反应导致关键的促癌和抑癌基因突变,胰腺细胞增殖不受控制,经历腺泡导管化生(acinar-to-ductal metaplasia, ADM)和胰腺上皮内瘤变(pancreatic intraepithelial neoplasia, PanIN)等过程,向侵袭性胰腺癌发展[7]。了解胰腺炎——癌转化的发生机制对胰腺癌的早期诊断和治疗有十分重要的意义。
表观遗传是指DNA序列不发生变化,但基因表达却发生了可遗传的改变,且这种改变在发育和细胞增殖过程中能稳定传递。主要包括:(1)基因选择性转录表达的调控:DNA甲基化、组蛋白修饰、染色质重塑;(2)基因转录后的调控:非编码的RNA如微小RNA(microRNA, miRNA)、长链非编码RNA(long non-coding RNA, lncRNA)等[8]。表观遗传学参与了肿瘤的起始、发展、侵袭和转移[9-10]。有研究发现,DNA甲基化、组蛋白修饰、非编码RNA的调控等表观遗传修饰参与了胰腺癌的发生和恶性表型的维持[11]。表观遗传修饰在胰腺炎——癌转化中也发挥了非常重要的作用,本文拟对DNA甲基化、组蛋白修饰和非编码RNA的调控等表观遗传修饰在胰腺炎——癌转化中的研究进展作一综述。
1 胰腺炎——癌转化过程中的生物学变化慢性胰腺炎发展为胰腺癌是一个长期的过程,慢性胰腺炎导致活性氧(reactive oxygen species, ROS)持续过量产生,ROS调节一系列的致癌信号通路,诱发致癌基因变化,包括基因突变、表观遗传改变和染色体重排[6]。ROS可以直接引起氧化性DNA损伤,诱发点突变。有分析[12]发现,在慢性胰腺炎持续3年以上才会出现KRAS突变,而在恶性程度逐渐升高的PanIN-1、PanIN-2至PanIN-3病变中,则出现36%、44%~87%的KRAS突变,这表明,在长期氧化应激过程中,点突变积累形成致癌性突变。此外,在慢性胰腺炎中,慢性氧化应激导致DNA错配修复(mis-match repair, MMR)的活性受损,DNA甲基转移酶(DNA methyltransferases, DNMTs)过度表达,长期暴露于氧化应激环境下导致DNA甲基化变化持续遗传,从而导致肿瘤抑制因子失活。另外,ROS可以导致端粒缩短,抑制端粒酶活性,增加染色体不稳定性,从而导致染色体重排,见图 1。

|
| 图 1 胰腺炎——癌转化过程中的生物学变化 Figure 1 Biological changes during pancreatitis-carcinoma transformation |
ADM是慢性胰腺炎导致胰腺损伤后的初始反应,也是慢性胰腺炎向胰腺癌转化的重要过程,NF-κB等多种信号通路与细胞因子参与调节了这一过程[13]。胰腺腺泡细胞可以依赖NF-κB信号通路释放细胞因子募集白细胞,而白细胞可以进一步刺激腺泡细胞放大NF-κB信号相关通路,造成胰腺损伤从而诱导细胞ADM。同时,去分化的腺泡细胞可以继续通过募集并激活炎性细胞促进NF-κB信号转导,从而形成正反馈回路。此外,EGFR、JAK-STAT等信号通路也可在慢性胰腺炎的情况下促进ADM。
2 DNA甲基化DNA甲基化是指CG二核苷酸序列(C-phosphodiester-G, CpG)的胞嘧啶核苷酸5'端在DNA甲基转移酶(DNA methyltransferase, DNMT)的催化作用下,以腺苷甲硫氨酸(S-adenosyl methionine, SAM)作为甲基供体,通过共价键结合的方式获得一个甲基基团的化学修饰过程。
2.1 细胞因子信号转导抑制因子3(SOCS3)DNA甲基化再生胰岛衍生蛋白3-alpha(regenerating islet derived protein 3 alpha, Reg3A)在胰腺炎中表达显著增加,同时在胰腺癌中也存在过表达[14]。细胞因子信号转导抑制因子3(suppressor of cytokine signaling 3, SOCS3)可以在Reg3A的下游发挥作用。SOCS3作为JAK/STAT3信号通路的负反馈抑制剂发挥肿瘤抑制功能,而甲基化使SOCS3在肿瘤中沉默也已经有了很多报道。因此,有学者推测,甲基化使SOCS3下调与Reg3A过表达一起促进炎性反应相关胰腺癌的发生,并对此进行了相关研究[15]。结果发现,DNA甲基化使SOCS3在胰腺癌细胞中被抑制,而使用DNA甲基化抑制剂恢复SOCS3表达后,胰腺癌细胞生长受抑制,凋亡水平增加。在36个胰腺癌患者标本中,SOCS3甲基化程度高,且与Reg3A过表达吻合;在五个胰腺癌细胞系中也同样观察到SOCS3抑制与Reg3A过表达。使用外源Reg3A刺激胰腺癌细胞系可促进细胞生长,再过表达SOCS3后,可发现细胞活力降低,生长受到抑制,这表明SOCS3过表达可对抗外源Reg3A对胰腺癌细胞生长的促进作用。敲低正常胰腺上皮细胞中的SOCS3后,外源Reg3A的促增殖作用明显增强,表明SOCS3下调在Reg3A介导的胰腺上皮细胞恶性转化中起关键作用。此外,对Reg3A介导的信号转导途径中SOCS3下游信号转导的进一步研究显示,JAK2/STAT3和NF-κB通路可能参与了信号转导过程,但具体的作用机制有待进一步研究。
综上所述,甲基化使SOCS3下调与Reg3A过表达一起,通过JAK/STAT3和NF-κB通路,促进炎性反应相关胰腺癌的发生。
2.2 癌高甲基化基因启动子甲基化癌高甲基化基因(hypermethylated in cancer 1, HIC1)是一种转录调节因子,在慢性肝炎进展为肝细胞癌的过程中,HIC1甲基化程度不断升高[16]。SIRT1是HIC1的直接靶基因,HIC1甲基化上调SIRT1在乳腺癌和肺癌的发生发展中发挥作用[17-18]。因此,Zhao等[19]推测慢性胰腺炎可能导致HIC1启动子高甲基化,影响HIC1/SIRT1途径,从而导致胰腺癌的发生。
进一步研究发现,慢性胰腺炎和胰腺癌中HIC1启动子甲基化程度较正常胰腺组织明显升高,HIC1表达明显降低,而SIRT1表达与HIC1呈负相关。使用去甲基化试剂逆转HIC1甲基化,HIC1恢复表达,而SIRT1表达则明显降低。在胰腺癌细胞中过表达HIC1,SIRT1表达明显下调,细胞生长受到抑制,细胞周期停滞,细胞凋亡增加;恢复SIRT1表达后,细胞生长的抑制和细胞周期的停滞得以恢复。
综上所述,慢性胰腺炎中的HIC1启动子高甲基化导致HIC1/SIRT1途径异常从而促进胰腺癌的发生。
2.3 多基因甲基化慢性胰腺炎是胰腺癌最重要的危险因素之一,慢性胰腺炎发展为胰腺癌通常需要10~20年[20],因此发现可以预测这一过程的生物标志可以为胰腺癌的早期诊断和治疗提供机会。研究发现,慢性胰腺炎、胰腺癌前病变以及胰腺癌的DNA甲基化谱存在明显差异,了解从慢性胰腺炎到侵袭性胰腺癌各阶段DNA甲基化的变化,可以帮助寻找预测胰腺炎——癌转化的生物标志物。
Natale等总结了近年来发现的在胰腺炎——癌转化中表现出DNA甲基化差异的基因[21]。ADAMTS1、CDKN2A、DAPK1、GSTP1、MGMT和SOCS1在慢性胰腺炎中未甲基化,而在胰腺上皮内瘤变或胰腺导管内乳头状黏液性瘤中则出现甲基化,因此可以作为胰腺炎转化为胰腺癌过程早期的生物标志物。其中CDKN2A甲基化只出现在低级别上皮内瘤变中,而其他5个基因则在进展为侵袭性胰腺癌时甲基化程度升高;除此之外,APC、BRCA1、CLDN5、LHX1、RPRM、SFRP1和TJR2在进展为侵袭性胰腺癌时也会出现甲基化程度升高,它们可作为胰腺癌侵袭性阶段的生物标志物,见表 1。
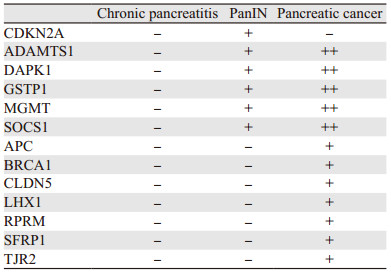
|
单一的DNA甲基化不足以区分慢性胰腺炎、胰腺癌前病变和胰腺癌,但聚集生物标志物则对这一过程有较强的预测能力。这不仅可以实现胰腺癌早期诊断,而且对肿瘤分期检测、个体化治疗等也有帮助。
3 组蛋白修饰组蛋白包括H1、H2A、H2B、H3和H4五种成分,其中H2A、H2B、H3和H4分别以二聚体相结合,与DNA共同组成染色质的基本单位核小体。组蛋白修饰是指组蛋白在相关酶作用下发生甲基化、乙酰化、泛素化等修饰的过程,其在胰腺癌发生发展中发挥了重要作用[13]。
3.1 组蛋白去乙酰化酶组蛋白去乙酰化酶(histone deacetylase, HDAC)参与了胰腺炎中的炎性反应、组织损伤和纤维化等致病过程,同时也在胰腺癌中高表达,参与了胰腺癌的发生发展。因此HDAC与胰腺炎——癌转化关系密切,HDAC抑制剂可在胰腺癌的治疗中发挥作用[22]。
Kaneta等[23]研究发现,胰腺的生长和维持需要E—钙黏蛋白(Cdh1),Cdh1缺失会导致胰腺炎性反应和纤维化,也会促进胰腺细胞癌变。同时,Cdh1缺失可以增加HDAC1的表达。因此,HDAC1可以作为Cdh1缺乏的胰腺癌的治疗靶点。
慢性胰腺炎中所有炎性细胞因子均与胰腺癌的发生有关,因此抗炎对于胰腺癌的治疗具有重要意义。我们已经知道,HDAC抑制剂可以改善炎性反应相关的癌症。Chehl等[24]既往的研究发现百里醌(thymoquinone, Tq)可以抑制HDAC活性,诱导组蛋白超乙酰化,从而抑制胰腺癌细胞生长,促进其凋亡,因此他们认为Tq在胰腺癌中可能具有抗炎作用。后续研究证实,Tq对慢性胰腺炎中的5种细胞因子或趋化因子(TNF-α、MCP-1、Cox-2、IL-8、IL-1β)有抑制作用,且随着Tq剂量和时间的增加,抑制作用增强,24 h后几乎可以完全抑制。NF-κB的激活是激活各种细胞/趋化因子的重要步骤,Tq可以通过抑制NF-κB信号转导途径或抑制其转录从而抑制NF-κB活性。因此,Tq可以抑制HDAC活性,在胰腺癌中发挥抗炎作用,NF-κB是Tq对各种炎性介质抑制作用的可能靶标。
3.2 组蛋白去乙酰化酶相关蛋白SIN3BSIN3B是一种非催化性的支架蛋白,可作为组蛋白去乙酰化酶HDAC1/2转录抑制复合物的成分。Rielland等[25]研究发现,SIN3B通过上调IL-1α的分泌,促进促炎性肿瘤微环境,从而促进胰腺病变的进展。
敲除SIN3B基因的小鼠,其由KRAS基因驱动的PanIN进展为PDAC的时间显著延长,生存时间也显著延长,这表明SIN3B可以促进KRAS驱动的癌症进展。KRAS基因表达后,通过激活ERK1/2、STAT3和NF-κB等途径介导肿瘤细胞分泌IL-1α等炎性因子,SIN3B缺失的小鼠,其ERK1/2及STAT3的激活明显降低,且IL-1α也明显降低,因此由Sin3B缺失引起的PanIN的延迟进展与炎性反应的明显损害有关。在人体组织标本中,可以观察到胰腺炎和PanIN中SIN3B的表达明显增加,高表达SIN3B的标本中的pSTAT3和IL-1α也明显增加。因此可以推测,KRAS基因激活后,SIN3B通过上调IL-1α的分泌,促进促炎性肿瘤微环境,从而促进胰腺病变的进展。IL-1α是急性胰腺炎后表达的中枢细胞因子,Rielland实验室的初步数据显示,IL-1α与胰腺炎导致的ADM呈正相关,由于急性胰腺炎反复发作可发展为慢性胰腺炎,这是众所周知的PDAC危险因素,因此可以通过使用SIN3B/HDAC1/2复合抑制剂抑制胰腺中IL-1α的产生,延长ADM和PanIN的进展,这可以作为胰腺癌治疗的新靶标。
4 非编码RNA的调控非编码RNA是指不编码蛋白质的RNA,具有转录后调控的作用[13]。lncRNA是长度大于200 nt的非编码RNA,主要通过染色质修饰、转录和转录后水平调节来影响基因表达。microRNA是长约22 nt的非编码RNA,具有转录后负调控基因表达的作用。lncRNA和microRNA在胰腺炎——癌转化中发挥了重要的调控作用。
4.1 lncRNA与胰腺炎——癌转化 4.1.1 lncRNA-NUTF2P3-001lncRNA-NUTF2P3-001在慢性胰腺炎和胰腺癌中均过表达。KRAS是胰腺癌最重要的启动子之一[26],lncRNA-NUTF2P3-001与胰腺癌中KRAS的表达呈正相关。下调lncRNA-NUTF2P3-001后,KRAS及其下游蛋白表达减少,同时胰腺癌细胞的增殖能力减弱,细胞活力降低;反之,lncRNA-NUTF2P3-001过表达会上调KRAS,促进癌细胞存活及侵袭。这说明lncRNA-NUTF2P3-001可能通过上调KRAS表达从而在胰腺癌的发生中发挥启动子作用。进一步研究发现,与慢性胰腺炎相比,胰腺癌中miR3923的表达显著下调,miR3923的抑制可上调KRAS及其下游蛋白表达,而lncRNA-NUTF2P3-001通过与miR3923竞争性结合抑制miR3923表达。因此,lncRNA-NUTF2P3-001可以通过与miR3923竞争性结合,抑制miR3923表达,从而上调KRAS通路,在胰腺癌中发挥启动子作用[27]。
4.1.2 lncRNA-ABHD11-AS1Liu等[28]筛选出11个lncRNA,检测其在15例健康受试者、15例慢性胰腺炎以及15例胰腺癌受试者血浆中的含量,结果发现,lncRNA-ABHD11-AS1的含量存在明显差异,且联合lncRNA-ABHD11-AS1与胰腺癌肿瘤标志物CA19-9对于胰腺癌的检测更有效。因此,lncRNA-ABHD11-AS1可以作为胰腺癌检测的潜在标志物。
4.2 miRNA与胰腺炎——癌转化 4.2.1 miR-217研究发现,miR-217-SIRT1通路参与胰腺炎——癌转化过程。上皮间充质转化(epithelial-mesenchymal transformation, EMT)在慢性炎性反应向癌症转化的过程中发挥了重要作用[29]。TGF-β1在慢性胰腺炎和胰腺癌中均过度表达,诱发EMT,促进肿瘤发生发展[30-31]。慢性胰腺炎和胰腺癌组织中miR-217显著降低,与其中活跃的EMT呈正相关。SIRT1是miR-217的直接靶标,miR-217通过抑制SIRT1表达,诱发腺泡导管化生。TGF-β1处理的胰腺癌细胞中的miR-217表达降低,SIRT1表达显著增加,且表现出明显的EMT;若恢复miR-217的表达,SIRT1的表达受到抑制,EMT过程则会被逆转。因此,TGF-β1调控miR-217-SIRT1通路,诱发EMT,参与胰腺癌的发生发展[32]。
4.2.2 miR-143慢性胰腺炎和胰腺癌中miR-143表达均明显下调,而上调miR-143可以抑制胰腺癌细胞增殖,诱导细胞凋亡。TGF-β1激活激酶1(TAK1)是miR-143的直接靶基因,miR-143下调诱导TAK1高度表达,调节胰腺癌的发生发展。胰腺癌患者中TAK1表达与其下游的MAPK和NF-κB的表达呈正相关,而miR-143可下调胰腺癌细胞中TAK1及其下游MAPK和NF-κB的表达。以上这些结果说明,miR-143下调诱导TAK1高表达,通过MAPK和NF-κB途径,调节胰腺癌的发生发展[33]。
表观遗传学在胰腺炎——癌转化中发挥了重要的作用,为胰腺炎——癌转化这一过程提供了可能的预测方法,也为胰腺癌的早期诊断和治疗提供了有潜力的靶点。目前已知的表观遗传修饰在胰腺炎——癌转化中的作用和机制比较有限,还需进一步研究和阐明。
作者贡献
岳铭、徐海燕:查阅文献、撰写及修改论文
张晓飞:选题、提供修改意见
王理伟:确定选题及指导文章写作
| [1] |
Bray F, Ferlay J, Soerjomataram I, et al. Global cancer statistics 2018: GLOBOCAN estimates of incidence and mortality worldwide for 36 cancers in 185 countries[J]. CA Cancer J Clin, 2018, 68(6): 394-424. DOI:10.3322/caac.21492 |
| [2] |
Zeng H, Chen W, Zheng R, et al. Changing cancer survival in China during 2003–15: a pooled analysis of 17 population-based cancer registries[J]. Lancet Glob Health, 2018, 6(5): e555-e567. DOI:10.1016/S2214-109X(18)30127-X |
| [3] |
Oberstein PE, Olive KP. Pancreatic cancer: why is it so hard to treat?[J]. Therap Adv Gastroenterol, 2013, 6(4): 321-337. DOI:10.1177/1756283X13478680 |
| [4] |
Stathis A, Moore MJ. Advanced pancreatic carcinoma: current treatment and future challenges[J]. Nat Revi Clin Oncol, 2010, 7(3): 163-172. DOI:10.1038/nrclinonc.2009.236 |
| [5] |
Martel CD, Ferlay J, Franceschi S, et al. Global burden of cancers attributable to infections in 2008: a review and synthetic analysis[J]. Lancet Oncol, 2012, 13(6): 607-615. DOI:10.1016/S1470-2045(12)70137-7 |
| [6] |
Dai JJ, Jiang MJ, Wang XP, et al. Inflammation-Related Pancreatic Carcinogenesis: Mechanisms and Clinical Potentials in Advances[J]. Pancreas, 2017, 46(8): 973-985. DOI:10.1097/MPA.0000000000000886 |
| [7] |
Zambirinis CP, Pushalkar S, Saxena D, et al. Pancreatic cancer, inflammation, and microbiome[J]. Cancer J, 2014, 20(3): 195-202. DOI:10.1097/PPO.0000000000000045 |
| [8] |
Dawson MA, Kouzarides T. Cancer Epigenetics: From Mechanism to Therapy[J]. Cell, 2012, 150(1): 12-27. DOI:10.1016/j.cell.2012.06.013 |
| [9] |
Werner RJ, Kelly AD, Issa JJ. Epigenetics and Precision Oncology[J]. Cancer J, 2017, 23(5): 262-269. DOI:10.1097/PPO.0000000000000281 |
| [10] |
Kanwal R, Gupta K, Gupta S, et al. Cancer Epigenetics: An Introduction[J]. Methods Mol Biol, 2015, 1238: 3-25. |
| [11] |
Neureiter D, Jäger T, Ocker M, et al. Epigenetics and pancreatic cancer: pathophysiology and novel treatment aspects[J]. World J Gastroenterol, 2014, 20(24): 7830-7848. DOI:10.3748/wjg.v20.i24.7830 |
| [12] |
Chang XY, Wu Y, Li Y, et al. Intraductal papillary mucinous neoplasms of the pancreas: Clinical association with KRAS[J]. Mol Med Rep, 2018, 17(6): 8061-8068. |
| [13] |
Murtaugh LC, Keefe MD. Regeneration and repair of the exocrine pancreas[J]. Annu Rev Physiol, 2015, 77: 229-249. DOI:10.1146/annurev-physiol-021014-071727 |
| [14] |
Zhang MY, Wang J, Guo J. Role of Regenerating Islet-Derived Protein 3A in Gastrointestinal Cancer[J]. Front Oncol, 2019, 9: 1449. DOI:10.3389/fonc.2019.01449 |
| [15] |
Wang J, Zhou H, Han Y, et al. SOCS3 methylation in synergy with Reg3A overexpression promotes cell growth in pancreatic cancer[J]. J Mol Med(Berl), 2014, 92(12): 1257-1269. |
| [16] |
Liu M, Jiang L, Guan XY. The genetic and epigenetic alterations in human hepatocellular carcinoma: a recent update[J]. Protein Cell, 2014, 5(9): 673-691. DOI:10.1007/s13238-014-0065-9 |
| [17] |
Chen WY, Wang DH, Yen RC, et al. Tumor Suppressor HIC1 Directly Regulates SIRT1 to Modulate p53-Dependent DNA-Damage Responses[J]. Cell, 2005, 123(3): 437-448. DOI:10.1016/j.cell.2005.08.011 |
| [18] |
Tseng RC, Lee CC, Hsu HS, et al. Distinct HIC1-SIRT1-p53 loop deregulation in lung squamous carcinoma and adenocarcinoma patients[J]. Neoplasia, 2009, 11(8): 763-770. DOI:10.1593/neo.09470 |
| [19] |
Zhao G, Qin Q, Zhang J, et al. Hypermethylation of HIC1 Promoter and Aberrant Expression of HIC1/SIRT1 Might Contribute to the Carcinogenesis of Pancreatic Cancer[J]. Ann Surg Oncol, 2013, 20(Suppl 3): S301-S311. |
| [20] |
Raimondi S, Lowenfels AB, Morselli-Labate AM, et al. Pancreatic cancer in chronic pancreatitis; aetiology, incidence, and early detection[J]. Best Pract Res Clin Gastroenterol, 2010, 24(3): 349-358. DOI:10.1016/j.bpg.2010.02.007 |
| [21] |
Natale F, Vivo M, Falco G, et al. Deciphering DNA methylation signatures of pancreatic cancer and pancreatitis[J]. Clin Epigenetics, 2019, 11(1): 132. DOI:10.1186/s13148-019-0728-8 |
| [22] |
Klieser E, Swierczynski S, Mayr C, et al. Role of histone deacetylases in pancreas: Implications for pathogenesis and therapy[J]. World J Gastrointest Oncol, 2015, 7(12): 473-483. DOI:10.4251/wjgo.v7.i12.473 |
| [23] |
Kaneta Y, Sato T, Hikiba Y, et al. Loss of Pancreatic E-Cadherin Causes Pancreatitis-Like Changes and Contributes to Carcinogenesis[J]. Cell Mol Gastroenterol Hepatol, 2020, 9(1): 105-119. DOI:10.1016/j.jcmgh.2019.09.001 |
| [24] |
Chehl N, Chipitsyna G, Gong Q, et al. Anti-inflammatory effects of the Nigella sativa seed extract, thymoquinone, in pancreatic cancer cells[J]. HPB(Oxford), 2009, 11(5): 373-381. |
| [25] |
Rielland M, Cantor DJ, Graveline R, et al. Senescence-associated SIN3B promotes inflammation and pancreatic cancer progression[J]. J Clin Invest, 2014, 124(5): 2125-2135. DOI:10.1172/JCI72619 |
| [26] |
Jones S, Zhang X, Parsons DW, et al. Core signaling pathways in human pancreatic cancers revealed by global genomic analyses[J]. Science, 2008, 321(5897): 1801-1806. DOI:10.1126/science.1164368 |
| [27] |
Li X, Deng SJ, Zhu S, et al. Hypoxia-induced lncRNA-NUTF2P3-001 contributes to tumorigenesis of pancreatic cancer by derepressing the miR-3923/KRAS pathway[J]. Oncotarget, 2016, 7(5): 6000-6014. DOI:10.18632/oncotarget.6830 |
| [28] |
Liu Y, Feng W, Liu W, et al. Circulating lncRNA ABHD11-AS1 serves as a biomarker for early pancreatic cancer diagnosis[J]. J Cancer, 2019, 10(16): 3746-3756. DOI:10.7150/jca.32052 |
| [29] |
Suarez-Carmona M, Lesage J, Cataldo D, et al. EMT and inflammation: inseparable actors of cancer progression[J]. Mol Oncol, 2017, 11(7): 805-823. DOI:10.1002/1878-0261.12095 |
| [30] |
Zhou Q, Xia S, Guo F, et al. Transforming growth factor-β in pancreatic diseases: Mechanisms and therapeutic potential[J]. Pharmacol Res, 2019, 142: 58-69. DOI:10.1016/j.phrs.2019.01.038 |
| [31] |
Miyazono K, Katsuno Y, Koinuma D, et al. Intracellular and extracellular TGF-β signaling in cancer: some recent topics[J]. Front Med, 2018, 12(4): 387-411. DOI:10.1007/s11684-018-0646-8 |
| [32] |
Deng S, Zhu S, Wang B, et al. Chronic pancreatitis and pancreatic cancer demonstrate active epithelial–mesenchymal transition profile, regulated by miR-217-SIRT1 pathway[J]. Cancer Lett, 2014, 355(2): 184-191. DOI:10.1016/j.canlet.2014.08.007 |
| [33] |
Huang FT, Peng JF, Cheng WJ, et al. MiR-143 Targeting TAK1 Attenuates Pancreatic Ductal Adenocarcinoma Progression via MAPK and NF-κB Pathway In Vitro[J]. Dig Dis Sci, 2017, 62(4): 944-957. DOI:10.1007/s10620-017-4472-7 |