 2021, Vol. 48
2021, Vol. 48文章信息
- 基于SEER数据库的儿童青少年室管膜瘤预后Nomogram模型的构建和验证
- Establishment and Validation of A Prognostic Nomogram for Pediatric Ependymoma Based on SEER Database
- 肿瘤防治研究, 2021, 48(4): 358-364
- Cancer Research on Prevention and Treatment, 2021, 48(4): 358-364
- http://www.zlfzyj.com/CN/10.3971/j.issn.1000-8578.2021.20.0954
- 收稿日期: 2020-08-17
- 修回日期: 2020-11-01
引用本文 |



2. 830011 乌鲁木齐,新疆医科大学公共卫生学院儿少卫生与妇幼保健教研室
2. Department of Child and Maternal Health, School of Public Health, Xinjiang Medical University, Urumqi 830011, China
室管膜瘤是一组起源于脑室或脊髓中央管室管膜细胞的神经胶质瘤,多见于儿童及青少年,约占儿童颅内肿瘤的10%,是儿童中枢神经系统的第三大肿瘤[1]。文献报道该病5年总生存率为50%~81%,无进展生存率为23%~69%,幸存者通常会经历多次肿瘤演进,并因高频的手术及放化疗而导致生活质量降低[1-3]。即使初诊时进行了肿瘤切除和放射治疗,仍有40%~50%的小儿患者会复发。儿童青少年室管膜瘤的复发多在原发部位,复发的患者预后较差,死亡风险高达90%[4-5]。患者的预后常与发病年龄、肿瘤原发部位、手术方式等因素密切相关。因儿童青少年室管膜瘤的稀有性,该类研究人群少,统计功效低,患者的预后预测研究常具有挑战性。以往儿童青少年室管膜瘤的研究基本局限于小样本或单一机构的临床数据,大型长期随访研究十分稀缺,针对儿童青少年群体的室管膜瘤预后预测模型也较为罕见[4, 6]。为提供可靠的预后生存评估,本文采用一项大型人群代表性癌症研究SEER数据库,建立并验证0~19岁儿童青少年人群的室管膜瘤预后预测模型。纳入较全面的社会人口学、临床和肿瘤相关变量并细化分类年龄组,个体化预测患者生存率,为临床治疗决策和临床研究方案的设计,提供预后预测指导。
1 资料与方法 1.1 一般资料本文数据来自美国国立癌症研究所的"监测、流行病学和结果(the Surveillance, Epidemiology, and End Results, SEER)数据库"。该库包含来自美国18个地理区域的数据,约占美国人口的28%,是现阶段北美大型肿瘤数据库之一。SEER数据库已取消患者信息识别,符合机构审查委员会和伦理委员会要求,信息可公开获取(https://seer.cancer.gov/data/options.html)。
1.2 样本纳入和剔除标准通过SEER*Stat软件8.3.8版本获取1975—2016年临床病理信息,软件可通过程序操作自动筛选合适的样本人群。在SEER*Stat中选择年龄范围、疾病代码,排除种族信息为"未知"者后初步得到1 371例。然后通过数据清理,剔除缺失放疗(20例)、手术方式(232例)以及扩散程度(2例)信息的患者,筛选流程见图 1。总共1 117例0~19岁患者被纳入本研究,其中896例被随机分为建模组,其余221例为验证组。

|
| 图 1 样本筛选流程图 Figure 1 Flow diagram of sample selection |
从SEER数据库中获取性别、种族、诊断年龄、原发部位、扩散程度、组织学分级、手术方式、放化疗情况、登记地点和生存时间等信息。因室管膜瘤的病理形态高度变异,无法用特定指标表现[7]。SEER数据库中对于肿瘤组织学的分类为:高分化,Ⅰ级(well differentiated, Grade Ⅰ);中度分化,Ⅱ级(moderately differentiated, Grade Ⅱ);分化不良,Ⅲ级(poorly differentiated, Grade Ⅲ);间变性,Ⅳ级(undifferentiated/anaplastic, Grade Ⅳ)以及分化情况不明(Unknown)。根据国际疾病分类(The International Classification of Diseases for Oncology, Third Edition, ICD-0-3)组织学代码9391、9392、9393和9394(室管膜瘤、间变性室管膜瘤、乳头型室管膜瘤和黏液乳头状室管膜瘤)作为病例纳入标准,临床死亡为研究结局。
1.3 统计学方法Mann-Whitney U检验比较两组的社会人口学和临床特征差异。Kaplan-Meier生存曲线比较入组患者不同年龄组总生存期。单变量和多变量Cox比例风险回归模型确定潜在的预测因素,基于生存分析结果构建Nomogram模型并验证,预测5年和10年总生存率。通过一致性指数(C-index),受试者工作特征曲线(ROC)和曲线下面积(AUC)值来评估列线图的辨别能力。C指数越高,预测价值越高。应用校准曲线以获取列线图预测的总体生存率与实际结果之间的一致性。对这些分析进行Bootstrap重复抽样(自抽样1 000次)。此外,通过决策曲线分析(DCA)评价列线图模型的临床实用性。所有统计分析均使用SEER*Stat软件,R(version 4.0.2)和Stata/SE 15.0(Stata Corporation, College Station, TX, USA),本研究采用双侧检验法,P < 0.05为差异有统计学意义。
2 结果 2.1 患者临床病理学特征建模组和验证组患者的社会人口学及临床病理基本特征见表 1。建模组患者的随访时间为0~493月,中位随访时间为60月。两组预测变量差异均无统计学意义,具有可比性(均P > 0.05)。不同年龄组总生存期Kaplan-Meier生存曲线比较见图 2(P < 0.001)。

|

|
| 图 2 不同年龄组Kaplan-Meier生存曲线 Figure 2 Kaplan-Meier curves of different age groups |
单因素和多因素Cox比例风险模型分析结果见表 2,诊断年龄、性别、种族、原发部位、组织学分级、手术方式和登记地点的差异有统计学意义。原发部位为脊髓的患者死亡风险为颅内原发患者的0.284倍,部分切除术患者的死亡风险为全切术患者的3.291倍。黑种人相对于白种人预后较差(HR=1.539, 95%CI: 0.896~2.644),但差异无统计学意义;API人群的死亡风险为白种人的2.186倍。与≤1岁患者相比,其他年龄组患者的预后良好。我们纳入登记地点以减少SEER数据库中因缺乏经济因素而造成的影响,结果表明登记地点Southeast的死亡风险是Northeast的2.196倍。
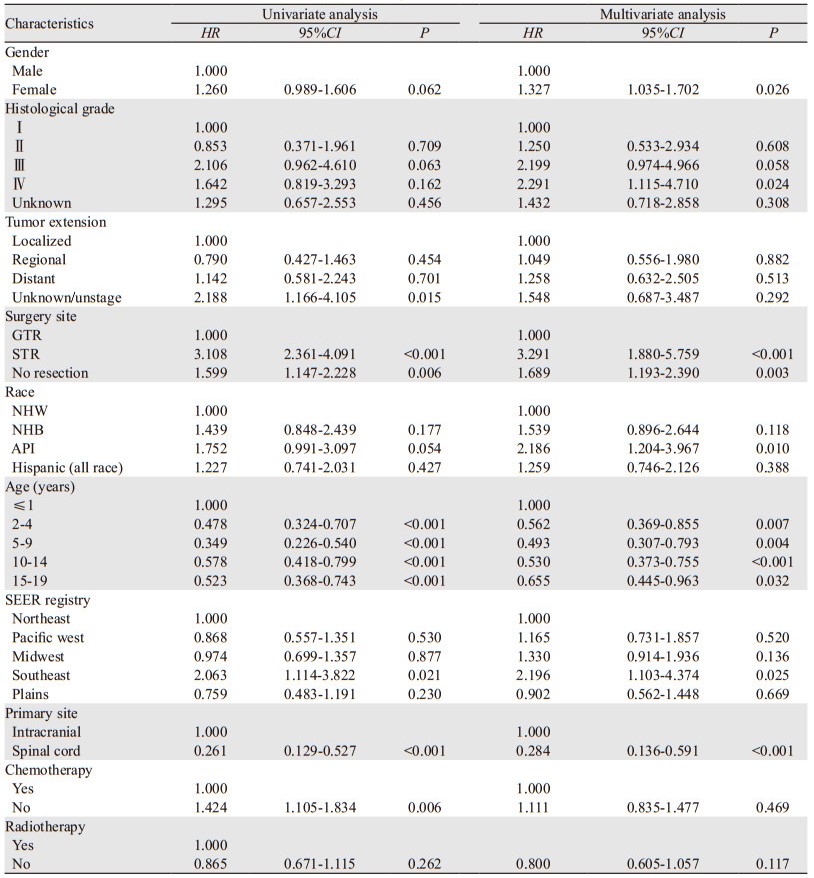
根据多因素Cox比例风险模型筛选出的差异有统计学意义的预后变量建立儿童青少年室管膜瘤风险列线图,包括诊断年龄、性别、种族、原发部位、组织学分级、手术方式和登记地点,根据每一项预后影响因素向上投映到分数尺上,再将各项分值相加对应到总分尺,从总分尺向下投射即可得出患者5年或10年的总生存率,见图 3。

|
| 图 3 建模组5年和10年总生存率列线图 Figure 3 Nomogram of 5- and 10-year overall survivals of training set |
验证结果显示,建模组的C指数为0.713(95% CI: 0.680~0.747),验证组C指数为0.734(95% CI: 0.681~0.787),认为模型区分能力尚可。建模组和验证组Nomogram预测模型5年、10年的曲线下面积(AUC)分别为0.741、0.722和0.703、0.687,认为模型准确性尚可,见图 4。建模组和验证组的校准曲线显示Nomogram模型与理想模型一致性尚可,见图 5。决策曲线分析显示在建模组和验证组中,风险阈值为0.20~0.40时,此Nomogram模型对5年和10年总生存率可表现出最佳净收益,见图 6。本研究的Nomogram模型在预测儿童青少年室管膜瘤患者的短期或长期总生存率方面比预后因素更好。

|
| 图 4 建模组和验证组5年(A, C)和10年(B, D)总生存率列线图的ROC曲线 Figure 4 ROC curves for predicting 5-(A, C) and 10-year (B, D) overall survivals of the nomogram in training set and validation set |

|
| 图 5 建模组和验证组5年(A, C)和10年(B, D)总生存率列线图的校准曲线 Figure 5 Calibration curves for predicting 5-(A, C) and 10-year(B, D) overall survivals of the nomogram in training set and validation set |

|
| 图 6 建模组和验证组5年(A, C)和10年(B, D)总生存率列线图的决策分析曲线 Figure 6 Decision curve analysis for predicting 5- (A, C) and 10-year(B, D) overall survivals of the Nomogram in training set and validation set |
儿童青少年室管膜瘤约占所有儿童脑和脊髓肿瘤的9%,美国每年约有0.29%的儿童青少年确诊[8]。根据细胞间变程度,世界卫生组织(WHO)神经系统肿瘤分类将室管膜瘤分为Ⅰ、Ⅱ和Ⅲ级(室管膜下瘤和黏膜乳头状室管膜瘤:WHOⅠ级;室间隔瘤:WHOⅡ级;RELA融合阳性室间隔膜瘤:WHOⅡ级或Ⅲ级;间变性室管膜瘤:WHOⅢ级)。80%的病例可局部复发,3%~9%可转移复发且恶性程度增高[9-10]。由于时间跨度以及肿瘤研究治疗的进步性,WHO分类系统会发生相应的变化,这会对组织学分级等记录的标准性产生影响,因此我们使用了基于SEER的肿瘤组织学分级。虽然这种分类方式会在一定程度上限制此模型的灵活性,但由于研究之间的分级标准差异以及对分级固有的主观性,在2015年召开的全球室管膜瘤共识会议上已经达成了组织学分级与结果之间并没有一致相关性的共识[11-13]。另外,提取数据的质量取决于数据输入SEER的方式,而其中的组织学和放射学结果尚未得到集中审查,室管膜瘤的分级可能存在很大差异[14]。
脑肿瘤根据组织学进行分类,但是肿瘤的位置、扩散程度、分子特征、年龄及手术切除范围是影响治疗和预后的重要因素[6, 15-16]。本研究结果与其相似。室管膜瘤的发病风险表现出双峰年龄分布,0~4岁时达到第一个峰值,在青少年早期下降,然后在30~50岁时出现第二个高峰[17]。本研究结果显示,婴幼儿室管膜瘤患者的死亡风险显著高于其他年龄组。由于分子生物学的异质性,低龄尤其是不能进行全切术和放射治疗的患者中,室管膜瘤的预后不良[4]。对于复发和低龄不宜放疗的患者,目前主要采用化学疗法[16, 18],但化学疗法在实施部分切除术和低龄儿童中的作用仍存在争议。一项针对儿童室管膜瘤的国际随机试验SIOP Ependymoma Ⅱ(欧洲)发现42%的3岁以下儿童采用术后化疗可达到5年无进展生存[19]。但另一项基于9 233例未成年儿童的研究结果显示,增加化疗剂量并未提高生存率,却显著增加了化疗不良反应发生率[20]。有研究显示儿童室管膜瘤的中位复发时间在诊断后2年内,因相关数据的缺失,本研究未能纳入肿瘤是否复发的信息[21]。我们还发现,API患者相对白种人患者表现出更高的死亡风险。有研究结果显示,API人群的中枢神经系统肿瘤的发病率与白种人相似[22]。API国外出生人口比例较高,社会经济水平,医疗资源获取率和族裔肿瘤生物学的差异可能会导致其生存率远低于白种人[23-24],另外,这些少数族裔对于放疗等辅助治疗的抵触也可能影响总生存率[25]。我们纳入"登记地点"以减少SEER数据库中因缺乏经济因素而造成的影响,结果显示登记地点Southeast的死亡风险高于Northeast,Southeast为美籍非裔聚集地,而黑种人具有相对更高的儿童室管膜瘤死亡风险[26-27],但我们的分析结果仅显示了更高的死亡风险但无统计学意义。
室管膜瘤的原发部位和治疗方式,尤其是手术切除方式被认为是影响肿瘤预后的指标[2, 16]。尽管有研究质疑,但手术全切辅助放疗仍是儿童室管膜瘤的首选治疗方案[11, 28]。部分切除术后放疗患者的总生存率为22%~52%,而全切术后放疗的患者总生存率可达67%~93%[1]。然而,如今大规模的基因组学和表观基因组学研究揭示了不同室管膜瘤分子亚组预后的差异性[29-30]。因此,当前和未来应全面评估不断发展的分子事件并开发合理有效的治疗靶标。良好的预后评估对室管膜瘤患者的治疗随访及后续治疗均有重要意义。Nomogram图能够直观地显示个体化风险评估。本研究利用常见临床病理变量建立了用于评估儿童青少年室管膜瘤5年和10年总生存率的预后模型Nomogram图,模型一致性和净受益尚可,对患者预后的个体化的预估有应用价值。
本研究也具有一定局限性。SEER数据库作为美国主要的肿瘤临床数据库,涉及人种多样,但主要为白种人和黑种人,亚洲人种的临床数据记录较少,除此之外,为减少经济因素对肿瘤预后的影响,利用了地理位置"登记地点"来代替原数据库中缺少的经济信息,因此该模型更适用于在美国居住的人群。SEER作为一个回顾性研究,具有无法避免的潜在选择偏倚,结果的普及性不理想。数据库中放疗和化疗信息不完整,也缺少肿瘤复发、临床症状、合并症和神经系统状况的相关记录。最后,根据美国对儿童青少年室管膜瘤人群的定义,我们纳入的研究对象为19岁以下人群,室管膜瘤低龄化患者预后差,因此可能会对研究结果产生影响。
综上所述,年龄、性别、种族、原发部位、组织学分级、手术方式及登记地点是儿童青少年室管膜瘤的预后影响因素,基于这些指标建立的风险预测模型准确性及临床应用性尚可,对医务工作者在临床工作中进行直观的、个体化的风险分析具有一定的参考价值,但本研究样本资料来源于美国癌症数据库,而种族易感性和地域差异对肿瘤发生率和生存率具有影响,在我国儿童青少年室管膜瘤的指导应用中需谨慎。未来仍需要加大儿童室管膜瘤研究工作的投入,建立大样本、多中心的研究对预后治疗提供更好的指导。
作者贡献:
朱迪:论文撰写和数据统计
张羽珊、郑守娟:文献收集和论文审校
王霞:论文审校
| [1] |
Vitanza NA, Partap S. Pediatric Ependymoma[J]. J Child Neurol, 2016, 31(12): 1354-1366. DOI:10.1177/0883073815610428 |
| [2] |
Eaton BR, Goldberg S, Tarbell NJ, et al. Long-term health-related quality of life in pediatric brain tumor survivors receiving proton radiotherapy at < 4 years of age[J]. Neuro Oncol, 2020, 22(9): 1379-1387. DOI:10.1093/neuonc/noaa042 |
| [3] |
van Iersel L, van Santen HM, Potter B, et al. Clinical impact of hypothalamic-pituitary disorders after conformal radiation therapy for pediatric low-grade glioma or ependymoma[J]. Pediatr Blood Cancer, 2020, 67(12): e28723. |
| [4] |
Ritzmann TA, Rogers HA, Paine SML, et al. A retrospective analysis of recurrent pediatric ependymoma reveals extremely poor survival and ineffectiveness of current treatments across central nervous system locations and molecular subgroups[J]. Pediatr Blood Cancer, 2020, 67(9): e28426. |
| [5] |
Board PPTE. Childhood soft tissue sarcoma treatment (PDQ?): Health Professional Version[EB/OL]. PDQ Cancer Information Summaries, Bethesda (MD), 2020[2020-12-03]. https://www.cancer.gov/types/soft-tissue-sarcoma/hp/child-soft-tissue-treatment-pdq.
|
| [6] |
Deng X, Zhang X, Yang L, et al. Personalizing age-specific survival prediction and risk stratification in intracranial grade Ⅱ/Ⅲ ependymoma[J]. Cancer Med, 2020, 9(2): 615-625. DOI:10.1002/cam4.2753 |
| [7] |
Duffner PK, Horowitz ME, Krischer JP, et al. Postoperative chemotherapy and delayed radiation in children less than three years of age with malignant brain tumors[J]. N Engl J Med, 1993, 328(24): 1725-1731. DOI:10.1056/NEJM199306173282401 |
| [8] |
Ostrom QT, Cioffi G, Gittleman H, et al. CBTRUS Statistical Report: Primary Brain and Other Central Nervous System Tumors Diagnosed in the United States in 2012-2016[J]. Neuro Oncol, 2019, 21(Suppl 5): v1-v100. |
| [9] |
Robertson PL, Zeltzer PM, Boyett JM, et al. Survival and prognostic factors following radiation therapy and chemotherapy for ependymomas in children: a report of the Children's Cancer Group[J]. J Neurosurg, 1998, 88(4): 695-703. DOI:10.3171/jns.1998.88.4.0695 |
| [10] |
Merchant TE, Li C, Xiong X, et al. Conformal radiotherapy after surgery for paediatric ependymoma: a prospective study[J]. Lancet Oncol, 2009, 10(3): 258-266. DOI:10.1016/S1470-2045(08)70342-5 |
| [11] |
Pajtler KW, Mack SC, Ramaswamy V, et al. The current consensus on the clinical management of intracranial ependymoma and its distinct molecular variants[J]. Acta Neuropathol, 2017, 133(1): 5-12. DOI:10.1007/s00401-016-1643-0 |
| [12] |
Ellison DW, Kocak M, Figarellaf-Branger D, et al. Histopathological grading of pediatric ependymoma: reproducibility and clinical relevance in European trial cohorts[J]. J Negat Results Biomed, 2011, 10(1): 7. DOI:10.1186/1477-5751-10-7 |
| [13] |
Godfraind C. Classification and controversies in pathology of ependymomas[J]. Childs Nerv Syst, 2009, 25(10): 1185-1193. DOI:10.1007/s00381-008-0804-4 |
| [14] |
Marinoff AE, Ma C, Guo D, et al. Rethinking childhood ependymoma: a retrospective, multi-center analysis reveals poor long-term overall survival[J]. J Neurooncol, 2017, 135(1): 201-211. DOI:10.1007/s11060-017-2568-8 |
| [15] |
Rudà R, Reifenberger G, Frappaz D, et al. EANO guidelines for the diagnosis and treatment of ependymal tumors[J]. Neuro Oncol, 2018, 20(4): 445-456. DOI:10.1093/neuonc/nox166 |
| [16] |
Merchant TE. Current Clinical Challenges in Childhood Ependymoma: A Focused Review[J]. J Clin Oncol, 2017, 35(21): 2364-2369. DOI:10.1200/JCO.2017.73.1265 |
| [17] |
Dorfer C, Tonn J, Rutka JT. Ependymoma: a heterogeneous tumor of uncertain origin and limited therapeutic options[J]. Handb Clin Neurol, 2016, 134: 417-431. |
| [18] |
Klish M, Shrieve DC, Macdonald DM, et al. Childhood ependymomas: An analysis of factors impacting survival[J]. Int J Radiat Oncol Biol Phys, 2004, 60(1): S309-S310. DOI:10.1016/j.ijrobp.2004.07.116 |
| [19] |
Grundy RG, Wiline SA, Weston CL, et al. Primary postoperative chemotherapy without radiotherapy for intracranial ependymoma in children: the UKCCSG/SIOP prospective study[J]. Lancet Oncol, 2007, 8(8): 696-705. DOI:10.1016/S1470-2045(07)70208-5 |
| [20] |
Strother DR, Lafay-Cousin L, Boyett JM, et al. Benefit from prolonged dose-intensive chemotherapy for infants with malignant brain tumors is restricted to patients with ependymoma: a report of the Pediatric Oncology Group randomized controlled trial 9233/34[J]. Neuro Oncol, 2014, 16(3): 457-465. DOI:10.1093/neuonc/not163 |
| [21] |
Mack SC, Taylor MD. Put away your microscopes: the ependymoma molecular era has begun[J]. Curr Opin Oncol, 2017, 29(6): 443-447. DOI:10.1097/CCO.0000000000000411 |
| [22] |
Achey RL, Vo S, Cioffi G, et al. Ependymoma, NOS and anaplastic ependymoma incidence and survival in the United States varies widely by patient and clinical characteristics, 2000-2016[J]. Neurooncol Pract, 2020, 7(5): 549-558. |
| [23] |
Hübner JM, Kool M, Pfister SM, et al. Epidemiology, molecular classification and WHO grading of ependymoma[J]. J Neurosurg Sci, 2018, 62(1): 46-50. |
| [24] |
Kehm RD, Spector LG, Poynter JN, et al. Does socioeconomic status account for racial and ethnic disparities in childhood cancer survival?[J]. Cancer, 2018, 124(20): 4090-4097. DOI:10.1002/cncr.31560 |
| [25] |
Patel CG, Stavas M, Perkins S, et al. Central Nervous System Disease, Education, and Race Impact Radiation Refusal in Pediatric Cancer Patients[J]. J Pediatr Hematol Oncol, 2017, 39(5): 382-387. DOI:10.1097/MPH.0000000000000843 |
| [26] |
Espey D, Paisano R, Cobb N. Regional patterns and trends in cancer mortality among American Indians and Alaska Natives, 1990-2001[J]. Cancer, 2005, 103(5): 1045-1053. DOI:10.1002/cncr.20876 |
| [27] |
Zhang C, Ostrom QT, Hansen HM, et al. European genetic ancestry associated with risk of childhood ependymoma[J]. Neuro Oncol, 2020, 22(11): 1637-1646. DOI:10.1093/neuonc/noaa130 |
| [28] |
Merchant TE, Bendel AE, Sabin ND, et al. Conformal Radiation Therapy for Pediatric Ependymoma, Chemotherapy for Incompletely Resected Ependymoma, and Observation for Completely Resected, Supratentorial Ependymoma[J]. J Clin Oncol, 2019, 37(12): 974-983. DOI:10.1200/JCO.18.01765 |
| [29] |
Khatua S, Mangum R, Bertrand KC, et al. Pediatric ependymoma: current treatment and newer therapeutic insights[J]. Future Oncol, 2018, 14(30): 3175-3186. DOI:10.2217/fon-2018-0502 |
| [30] |
Wang J, Sun C, Liu M, et al. The potentially therapeutic targets of pediatric anaplastic ependymoma by transcriptome profiling[J]. Neoplasma, 2020, 200529N. |