 2020, Vol. 47
2020, Vol. 47文章信息
- 结肠癌淋巴结转移的风险基因及列线图预测模型的构建
- Risk Genes and Nomogram Model for Lymph Node Metastasis of Colon Cancer
- 肿瘤防治研究, 2020, 47(12): 947-952
- Cancer Research on Prevention and Treatment, 2020, 47(12): 947-952
- http://www.zlfzyj.com/CN/10.3971/j.issn.1000-8578.2020.20.0234
- 收稿日期: 2020-03-20
- 修回日期: 2020-09-27
引用本文 |



结肠癌是常见的消化道恶性肿瘤之一,全球范围内结直肠癌发病率和死亡率分别居恶性肿瘤的第3位和第2位,结肠癌的5年总生存率约60%[1-3]。淋巴结转移作为肿瘤分期的重要依据,对结肠癌患者预后的影响已被多项研究报道[4-6]。了解淋巴结转移状态可以为评估预后、指导治疗等提供信息。手术病理诊断是评估淋巴结转移的主要手段。NCCN指南指出术中清扫淋巴结12个以上才能保证淋巴结状态的判断准确[7];对于不能手术或淋巴结清扫不充分的患者,淋巴结状态判断可能不准确。影像学也是了解淋巴结转移的主要手段,但由于判断的主观性等限制,其判断有一定偏倚,且其诊断敏感度可能弱于特异性[8]。
随着基因测序技术的发展,从基因及分子的角度了解疾病更有助于推动疾病的精准治疗。肿瘤组织的基因表达值高低由客观数值界定,避免了主观偏倚。本研究旨在从基因层面寻找结肠癌淋巴结转移的相关基因,并构建由基因组成的nomogram预测模型,帮助评估患者的淋巴结状态。
1 资料与方法 1.1 基因表达数据下载及过滤从癌症基因组图谱(The Cancer Genome Atlas,TCGA,https://portal.gdc.cancer.gov/)数据库下载获得结肠癌患者的基因测序数据。纳入标准:(1)非细胞株或动物来源样本;(2)经病理确诊为结肠癌;(3)临床信息包含明确的病理N分期;(4)诊断为N0期的患者需淋巴结清扫数目≥12个。不同探针对应相同基因名时,取平均值进行后续分析,并剔除低表达基因(在50%及以上的样本中FPKM表达量为0的基因),以保证纳入分析的基因有足够的表达量。
1.2 差异表达基因分析及通路富集分析经以上标准过滤后,TCGA基因集被用于差异表达基因(differentially expressed gene, DEG)分析,分析结果中FDR(false positive rate) < 0.05且在淋巴结阳性和阴性组间表达均值差异大于两倍(log2(fold change) > 1)的基因被认为有差异表达。使用GSEA方法[9]对淋巴阳性和阴性组进行基因集通路富集分析,鉴定两组在GO[10]功能集及KEGG[11]上显著富集的通路,富集显著的标准设定为FDR < 0.05。
1.3 淋巴结转移的风险基因鉴定和模型建立LASSO回归[12]对差异表达基因进一步降维,以AUC(area under receiver operating characteristic curve)值为标准,筛选转移风险关键基因。LASSO回归基于L1正则化方法对样本数据进行变量选择,将原本很小的系数直接压缩至0,从而将这部分系数所对应的变量视为非显著性变量,将不显著的变量直接舍弃。其原理主要基于L1正则化,是避免模型过拟合,将高维数据降维的有效方法。得到关键基因后利用赤池信息准则(Akaike information criterion, AIC)构建最优的logistics列线图(nomogram)模型。
1.4 模型的评估和验证采用ROC曲线及校准曲线对模型的表现力进行评估,同时对模型进行Hosmer-Lemeshow拟合优度检验。从NCBI平台下的GEO数据库下载外部验证数据。AUC(C统计量)大于0.65为理想模型。
1.5 决策曲线分析决策曲线分析法[13-14](decision curve analysis, DCA)用于探究在不同阳性阈值下,该模型对临床净获益率的影响。DCA横坐标为阈概率(threshold probability)。当nomogram模型评估值达到某值时,患者i的淋巴结转移概率记为Pi;当Pi达到某阈值(记为Pt),认定为阳性。DCA的纵坐标为净获益率(net benefit, NB), 定义为[真阳性数-假阳性数×Pt/(1-Pt)]/样本数,由NB对Pt作图可得DCA曲线。
1.6 统计学方法差异表达基因分析采用非配对Wilcox检验,差异表达基因分析及富集分析中的P值经过BH(Benjamin and Hochberg)法校正得到FDR值。模型变量的共线性通过方差膨胀因子(variance inflation factor, VIF)评估,VIF > 4时认为变量间存在多重共线性。所有统计分析均在R软件(https://www.r-project.org/)中实现,除差异表达分析及富集分析外,双侧P < 0.05为差异有统计学意义。
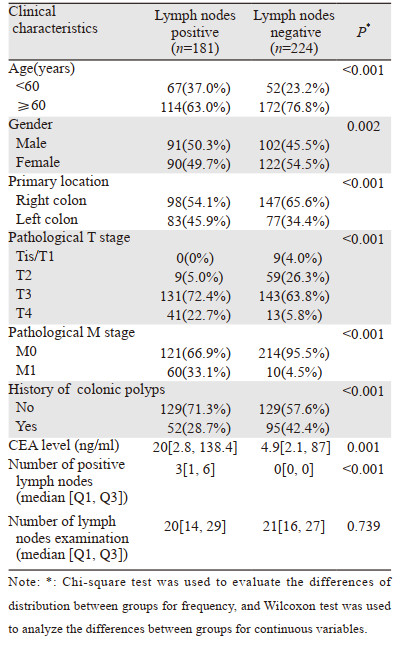
2 结果 2.1 患者基本信息经筛选,共纳入TCGA数据库中405例结肠癌患者的样本,其中未发生淋巴结转移的患者224例,发生淋巴结转移的患者181例,基本情况见表 1。在淋巴结阳性组中,< 60岁、男性、原发部位为左半结肠的患者较阴性组所占比例更高,淋巴结阴性组中,低T分期(Tis/T1/T2),低M分期(M0)及结肠息肉病史的患者所占比例更高。同时,淋巴结阳性组的CEA水平在统计学上显著高于淋巴结阴性组患者。两组淋巴结清扫数目无显著差别。外部验证数据来源于法国的多中心临床数据集(储存于GEO数据库中,数据集编号GSE39582),其中未发生淋巴结转移的患者312例,发生淋巴结转移的患者241例。
差异表达分析共鉴定差异基因55个,其中阳性组上调基因46个,下调基因9个,见图 1A~B。GSEA富集分析得到与淋巴结转移显著正相关的GO通路551条,包括介导细胞间信号传递的表面受体通路、细胞发育、细胞突起形成、细胞膜形态改变等;负相关的GO通路45条,包括肿瘤细胞免疫、黏膜免疫、细胞杀伤等;正相关的KEGG富集通路28条,包括黏着斑、PI3K-Akt信号通路、细胞外基质(ECM)受体相互作用、PPAR信号通路等;负相关KEGG通路38条,包括IL-17信号通路、Toll样受体信号通路、PD1/PD-L1表达调控通路及抗原呈递等,见图 1C~D。

|
| A: The heat map of differentially expressed genes, 46 genes were up-regulated and 9 genes were down-regulated; B: The volcano map of differentially expressed genes. The vertical axis is log10(FDR) which is the corrected P value. The horizontal axis is the logarithmic value of fold change between the two groups. The genes downregulated were in red, and those upregulated were in yellow, while non-changed genes were in blue; C, D: The enrichment analysis of GSEA based on KEGG database and GO database. The pathways shown in this figure met the criteria of FDR < 0.05. 图 1 差异表达基因及GESA功能富集分析 Figure 1 Differentially expressed genes and GESA enrichment analysis |
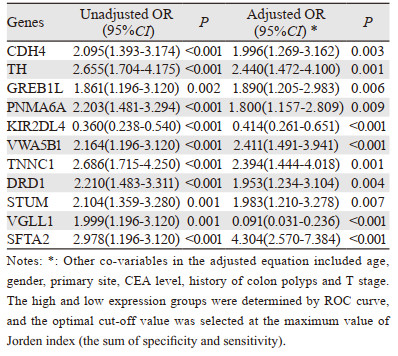
将编码基因进一步进行LASSO回归降维,共得到11个能有效预测淋巴结转移的基因:TH、GREB1L、VWA5B1、PNMA6A、TNNC1、KIR2DL4、DRD1、STUM、SFTA2、CDH4和VGLL1,见图 2。对TCGA数据集中上述11个基因的高低表达进行单因素及多因素logistic回归分析得到淋巴结转移的OR值,见表 2。校正前后的OR值显示CDH4、TH、GREB1L、PNMA6A、DRD1、VWA5B1、TNNC1、STUM、SFTA2的高表达提示淋巴结转移的风险增大,KIR2DL4的高表达提示淋巴结转移风险减小。VGLL1高表达在单因素分析中提示淋巴结转移的风险增大,而在多因素分析中提示淋巴结转移的风险减小。

|
| The AUC value was used as the standard to screen the key genes. A: The results without cross validation; B: The results after cross validation. 图 2 LASSO回归法筛选关键基因 Figure 2 Key genes screened by LASSO regression |
|
根据逐步回归分析的结果,由年龄、病理T分期、TH、CDH4、PNMA6A、TNNC1、KIR2DL4、STUM、SFTA2构成的模型具有最小AIC(440.4)值。进一步建立由上述变量组成的nomogram模型,见图 3,根据某患者每个变量实际情况找到其对应的刻度,向上投射到顶部的标尺(Points)即可得出每个变量的分值,分值相加即为总分值(total points),根据总得分值向下投射即可得到该患者的淋巴结转移风险概率(risk probability)。

|
| 图 3 Nomogram预测淋巴结转移模型 Figure 3 Nomogram model for predicting lymph node metastasis |
得到的模型进行内部验证发现模型的AUC值(AUC=0.800)、校准度及拟合优度(HL检验P > 0.05)均较佳,见图 4A~B,提示模型能很好地预测淋巴结转移风险。我们同时比较了纳入或不纳入基因的模型表现力,发现纳入基因后模型的AUC值得到了较大的提高。在TCGA和外部验证数据集中,AUC值的提高具有统计学意义(P < 0.001)。进一步验证发现,模型在法国多中心临床数据集上的AUC值、校准度及拟合优度均较佳,说明风险基因模型的外部数据适用性良好,见图 4C~D。

|
| A, B: The ROC and calibration curves of the model with genes and model without genes in the TCGA cohort; C, D: The ROC and calibration curves of the model with genes and model without genes in the validation cohort. The AUC values were presented together with ROC curves. And Hosmer-Lemeshow test was done when calculating calibration curves. For calibration curves, the solid line shows the ideal model which means the prediction probability is completely consistent with the actual rate. Dotted line represents the performance of genetic model, showing a good accuracy close to the ideal model between the rates of probability predicted by nomogram and the actual ones; E, F: The decision analysis curve of the model in TCGA and validation data sets. 图 4 Nomogram模型的内外部验证 Figure 4 I Internal and external validation of nomogram model |
决策曲线分析显示模型在TCGA数据集及外部验证数据集中,风险阈值为0.2~0.7时,基于含风险基因的nomogram模型进行干预决策带来的临床获益明显高于未考虑风险基因的模型,见图 4E~F。
3 讨论近年来,nomogram模型在癌症领域中的应用逐步增加,其将影响患者发病、预后或复发的各种临床病理或基因等多个因素纳入预测模型并可视化,将风险比量化为具体分值,通过简单运算即可获得预测疾病复发、转移及预后等的风险概率,为临床医生和研究者提供了方便有利的工具[15-17]。本研究模型验证结果提示其在预测淋巴结转移状态时有较好的一致性和区分度。GEO数据集上的验证结果进一步说明nomogram有良好的外部适用性。此外,决策曲线分析法表明使用nomogram评估可以在一定风险阈值下带来更高的临床获益。
淋巴结转移对结肠癌预后的重要影响已被多项研究报道,伴有淋巴结转移的患者较淋巴结阴性的患者生存更差,复发率也更高[18]。许多临床病理因素可能对淋巴结转移状态有重要影响。Yamaok等[19]发现,随着结肠癌浸润深度的增加,淋巴结转移率也逐渐升高。易小龙等[20]也发现肿瘤浸润深度与分化程度是影响结肠癌淋巴结转移的独立因素。值得注意的是,在我们的研究中,< 60岁组患者的淋巴结转移率高于≥60岁组,这可能与两组人群具有不同的临床特征有关[21]。影像学判断结肠癌淋巴结转移与否具有一定的局限性,通过联合其他指标可进一步提高术前诊断淋巴结转移的准确性。因此,本研究的nomogram模型可进一步协助临床医生判断淋巴结转移情况。
由于基因间可能存在很强的共线性,且考虑到结局事件数(淋巴结转移数)远大于自变量数量,本研究先采用LASSO回归进行降维,寻找差异基因中与淋巴结转移最为相关的基因。当一组基因高度相关时,LASSO回归会选出其中一个基因并且将其他因子收缩为零。LASSO回归最终鉴定了11个与淋巴结转移状态高度相关的蛋白编码基因。单、多因素分析提示仅KIR2DL4基因的高表达可能与抑制淋巴结转移有关。KIR2DL4基因编码杀伤细胞免疫球蛋白受体(killer-cell immunoglobulin-like receptor, KIR),在自然杀伤(natural killing, NK)细胞表面和少数T淋巴细胞表面表达,可识别并结合HLA-G分子,从而发挥对NK细胞的免疫抑制或激活的双重作用[22]。值得注意的是,CDH4基因编码的R-Cadherin蛋白(钙黏蛋白家族成员之一),其功能在不同肿瘤中可能存在异质性。据目前的研究报道,其可抑制鼻咽癌细胞的增殖和侵袭[23],但在骨肉瘤和胶质母细胞瘤中表现为促进增殖和侵袭转移的作用[24-25]。本研究中,CDH4的表达与结肠癌的淋巴结转移呈正相关。
与以往研究相比,本研究具有一定的优势。首先,以往的研究仅仅关注了临床病理因素[21, 26],这使淋巴结转移的预测效果有限。而本研究中的模型加入基因后,模型的诊断预测能力显著提高;其次,如前所述,影像学可以用于术前淋巴结转移与否的判断,但其主观性较强,而本研究中的模型基于基因表达量进行风险评分,可以有效避免主观判断的偏倚;最后,本研究中的模型使用更加方便,nomogram模型将预测的风险值量化,临床工作者仅需通过简单的计算即可获得确定的淋巴结转移概率值。但是,本研究也具有一定局限性,由于篇幅有限,对于11个风险基因的具体生物学功能有待进一步探究,且模型的适用性需在更大数据范围内进行验证。
综上所述,本研究鉴定出了11个与淋巴结转移相关的风险基因,并构建了由基因表达和临床因素组成的nomogram预测模型。经内部验证及外部验证发现,该模型有良好的区分度和校准度,并且可以带来潜在的临床获益。
作者贡献
武杰、宋启斌:论文撰写、数据统计
李岚、张惠博、吴思怡:论文撰写、文献收集
| [1] |
Miller KD, Nogueira L, Mariotto AB, et al. Cancer treatment and survivorship statistics, 2019[J]. CA Cancer J Clin, 2019, 69(5): 363-385. |
| [2] |
Bray F, Ferlay J, Soerjomataram I, et al. Global cancer statistics 2018: GLOBOCAN estimates of incidence and mortality worldwide for 36 cancers in 185 countries[J]. CA Cancer J Clin, 2018, 68(6): 394-424. |
| [3] |
Siegel RL, Miller KD, Fedewa SA, et al. Colorectal cancer statistics, 2017[J]. CA Cancer J Clin, 2017, 67(3): 177-193. |
| [4] |
Lykke J, Rosenberg J, Jess P, et al. Lymph node yield and tumour subsite are associated with survival in stage Ⅰ-Ⅲ colon cancer: results from a national cohort study[J]. World J Surg Oncol, 2019, 17(1): 62. |
| [5] |
Fortea-Sanchis C, Martínez-Ramos D, Escrig-Sos J. The lymph node status as a prognostic factor in colon cancer: comparative population study of classifications using the logarithm of the ratio between metastatic and nonmetastatic nodes (LODDS) versus the pN-TNM classification and ganglion ratio systems[J]. BMC Cancer, 2018, 18(1): 1208. |
| [6] |
Amri R, Klos CL, Bordeianou L, et al. The prognostic value of lymph node ratio in colon cancer is independent of resection length[J]. Am J Surg, 2016, 212(2): 251-257. |
| [7] |
Märkl B. Stage migration vs immunology: The lymph node count story in colon cancer[J]. World J Gastroenterol, 2015, 21(43): 12218-12233. |
| [8] |
Rollvén E, Blomqvist L, Öistämö E, et al. Morphological predictors for lymph node metastases on computed tomography in colon cancer[J]. Abdom Radiol (NY), 2019, 44(5): 1712-1721. |
| [9] |
Subramanian A, Tamayo P, Mootha VK, et al. Gene set enrichment analysis: a knowledge-based approach for interpreting genome-wide expression profiles[J]. Proc Natl Acad Sci U S A, 2005, 102(43): 15545-15550. |
| [10] |
Ashburner M, Ball CA, Blake JA, et al. Gene ontology: tool for the unification of biology. The Gene Ontology Consortium[J]. Nat Genet, 2000, 25(1): 25-29. |
| [11] |
Kanehisa M, Goto S, Sato Y, et al. KEGG for integration and interpretation of large-scale molecular data sets[J]. Nucleic Acids Res, 2012, 40(Database issue): D109-D114. |
| [12] |
Sauerbrei W, Royston P, Binder H. Selection of important variables and determination of functional form for continuous predictors in multivariable model building[J]. Stat Med, 2007, 26(30): 5512-5528. |
| [13] |
Fitzgerald M, Saville BR, Lewis RJ. Decision curve analysis[J]. JAMA, 2015, 313(4): 409-410. |
| [14] |
Vickers AJ, Elkin EB. Decision curve analysis: a novel method for evaluating prediction models[J]. Med Decis Making, 2006, 26(6): 565-574. |
| [15] |
Balachandran VP, Gonen M, Smith JJ, et al. Nomograms in oncology: more than meets the eye[J]. Lancet Oncol, 2015, 16(4): e173-e180. |
| [16] |
刘晓燕, 汤海涛, 王修中, 等. 早期胃癌患者ESD术后并发肺炎相关因素分析及风险模型预测[J]. 肿瘤防治研究, 2019, 46(10): 916-920. [Liu XY, Tang HT, Wang XZ, et al. Related Factors and Risk Model Prediction of Pneumonia After ESD in Patients with Early Gastric Cancer[J]. Zhong Liu Fang Zhi Yan Jiu, 2019, 46(10): 916-920.] |
| [17] |
庄金满, 卢婉婷, 黄玉秀, 等. 早期宫颈癌淋巴结转移的高危因素分析及列线图预测模型的构建[J]. 肿瘤防治研究, 2019, 46(1): 50-54. [Zhuang JM, Lu WT, Huang YX, et al. Risk Factors of Lymph Node Metastasis in Early-stage Cervical Cancer Patients and Build of A Nomogram Prediction Model[J]. Zhong Liu Fang Zhi Yan Jiu, 2019, 46(1): 50-54.] |
| [18] |
Böckelman C, Engelmann BE, Kaprio T, et al. Risk of recurrence in patients with colon cancer stage Ⅱ and Ⅲ: a systematic review and meta-analysis of recent literature[J]. Acta Oncol, 2015, 54(1): 5-16. |
| [19] |
Yamaoka Y, Kinugasa Y, Shiomi A, et al. The distribution of lymph node metastases and their size in colon cancer[J]. Langenbecks Arch Surg, 2017, 402(8): 1213-1221. |
| [20] |
易小龙, 李卫东. 结肠癌淋巴结转移的多因素回归分析[J]. 天津医科大学学报, 2010, 16(1): 78-80. [Yi XL, Li WD. Multivariate regression analysis of lymphatic metastasis in patients with colon carcinoma[J]. Tianjin Yi Ke Da Xue Xue Bao, 2010, 16(1): 78-80.] |
| [21] |
Xie X, Yin J, Zhou Z, et al. Young age increases the risk for lymph node metastasis in patients with early colon cancer[J]. BMC Cancer, 2019, 19(1): 803. |
| [22] |
Banerjee PP, Pang L, Soldan SS, et al. KIR2DL4-HLAG interaction at human NK cell-oligodendrocyte interfaces regulates IFN-gamma-mediated effects[J]. Mol Immunol, 2019, 115: 39-55. |
| [23] |
Du C, Huang T, Sun D, et al. CDH4 as a novel putative tumor suppressor gene epigenetically silenced by promoter hypermethylation in nasopharyngeal carcinoma[J]. Cancer Lett, 2011, 309(1): 54-61. |
| [24] |
Tang Q, Lu J, Zou C, et al. CDH4 is a novel determinant of osteosarcoma tumorigenesis and metastasis[J]. Oncogene, 2018, 37(27): 3617-3630. |
| [25] |
Ceresa D, Alessandrini F, Bosio L, et al. Cdh4 Down-Regulation Impairs in Vivo Infiltration and Malignancy in Patients Derived Glioblastoma Cells[J]. Int J Mol Sci, 2019, 20(16): 4028. |
| [26] |
Yan Y, Liu H, Mao K, et al. Novel nomograms to predict lymph node metastasis and liver metastasis in patients with early colon carcinoma[J]. J Transl Med, 2019, 17(1): 193. |