 2019, Vol. 46
2019, Vol. 46文章信息
- 第三代EGFR-TKIs治疗晚期非小细胞肺癌的耐药机制及应对策略研究进展
- Research Progress on Resistance Mechanisms and Overcoming Strategies to Third-generation EGFR-TKIs in Advanced Non-small Cell Lung Cancer
- 肿瘤防治研究, 2019, 46(10): 938-945
- Cancer Research on Prevention and Treatment, 2019, 46(10): 938-945
- http://www.zlfzyj.com/CN/10.3971/j.issn.1000-8578.2019.19.0169
- 收稿日期: 2019-02-15
- 修回日期: 2019-07-22
引用本文 |



肺癌是全世界癌症相关死亡的主要原因,非小细胞肺癌(non-small cell lung cancer, NSCLC)约占肺癌的85%[1]。大多数肺癌患者在诊断时已经是局部晚期或晚期,失去了手术治疗的机会,5年生存率仅为18%[2]。对于无法手术的晚期肺癌患者,全身治疗的主要手段为化疗、靶向治疗和免疫治疗。表皮生长因子受体(epidermal growth factor receptor, EGFR)基因是NSCLC中最常见的驱动基因之一。中国大陆地区NSCLC患者EGFR基因突变比例约为30%,其中女性、腺癌、非吸烟者的突变比例更高[1]。对于EGFR突变阳性的患者,EGFR酪氨酸激酶抑制剂(tyrosine kinase inhibitors, TKIs)的疗效明显优于传统化疗,国内外专业指南都建议将EGFR-TKIs作为EGFR敏感突变患者的一线治疗药物。然而,大多数患者服用一代或二代EGFR-TKIs 9~14月后会产生耐药,EGFR的20号外显子T790M突变是最常见机制,发生率高达50%[3]。以奥西替尼为代表的第三代EGFR-TKI用于克服T790M突变介导的获得性耐药取得了显著疗效。AURA3研究[3]表明奥西替尼二线治疗第一、二代EGFR-TKIs耐药后出现T790M突变的晚期NSCLC患者的中位无进展生存期(median progression-free survival, mPFS)长于含铂双药化疗。而FLAURA研究[4]表明奥西替尼一线治疗EGFR突变的晚期NSCLC的mPFS长于第一代EGFR-TKIs,奥西替尼可降低T790M突变NSCLC患者中枢神经系统进展的风险。
除奥西替尼外,针对T790M突变的第三代EGFR-TKIs还有Rociletinib、Olmutinib、Nazartinib和Naquotinib等。然而,患者在一线使用第三代EGFR-TKIs 18.9月,二线使用约10月后会再次出现耐药[3-4]。第三代EGFR-TKIs的耐药机制主要分为原发性耐药和获得性耐药,见图 1。目前对它的耐药机制报道较少,临床处理比较棘手。本文复习近三年的主要文献对第三代EGFR-TKIs治疗晚期NSCLC耐药机制及应对策略的研究进展进行综述。

|
| 图 1 第三代EGFR-TKIs治疗晚期EGFR突变NSCLC的原发性和获得性耐药机制 Figure 1 Primary and acquired resistance mechanisms of advanced EGFR-mutated NSCLC to the third-generation EGFR-TKIs |
原发性耐药的机制目前并不是十分明确,相关研究较少,可能机制包括BIM缺失多态性和EGFR 20外显子插入突变等,这些耐药机制导致EGFR突变患者对各代EGFR-TKIs的客观有效率并不能达到100%。
1.1 BIM缺失多态性BIM是B细胞淋巴瘤-2(B-cell lymphoma 2, BCL-2)家族成员之一,是一种促凋亡分子。BIM缺失多态性存在于约21%的东亚人中,在非洲和欧洲人群中不存在[5]。周彩存教授团队的一项研究显示,BIM缺失多态性是EGFR突变的NSCLC患者的不良预后因素,参与研究的352例NSCLC患者中有45例(12.8%)存在BIM缺失多态性,这部分患者使用第一代EGFR-TKIs治疗的mPFS仅为4.7月[6]。Tanimoto等通过体外研究发现,具有BIM缺失多态性的EGFR突变NSCLC细胞对第三代EGFR-TKI具有耐药性[7]。近期报道了一例EGFR L858R和T790M突变的NSCLC患者使用奥西替尼治疗的效果不佳,在外周血基因测序中发现存在BIM缺失多态性,且不伴有其他耐药突变。这一结果提示BIM缺失多态性也许可以作为预测NSCLC患者奥西替尼治疗疗效的生物标志物[5]。
1.2 EGFR 20外显子插入突变EGFR 20外显子插入突变在肺腺癌患者中的发生率约为3%,占所有EGFR突变的10%~12%[8],它可以阻滞EGFR-TKI与EGFR靶部位结合,导致原发性耐药。对于EGFR 20外显子插入突变的患者,一线使用一、二代EGFR-TKIs治疗的总有效率为3%~8%,mPFS仅为2月[9]。但第三代EGFR-TKIs治疗EGFR 20外显子插入突变患者的疗效缺乏相关报道。最近的一项体外研究显示,稳定表达EGFR 20外显子插入突变的细胞系对第三代EGFR-TKI具有耐药性[9]。表明EGFR 20外显子插入突变可能是第三代EGFR-TKIs的原发性耐药机制之一。目前奥西替尼用于EGFR 20外显子插入突变的NSCLC的Ⅱ期临床研究正在招募中(NCT03414814和NCT03191149)。
2 继发性耐药机制继发性耐药是指对药物敏感的肿瘤细胞在接触药物后通过改变自身代谢途径逃避药物的影响。第三代EGFR-TKIs的继发性耐药机制主要包括EGFR信号通路改变、旁路和下游信号通路异常激活、组织学转化等。AURA3[3]和FLAURA[4]研究中奥西替尼作为二线及一线治疗的继发性耐药机制,见图 2[10-11]。

EGFR 20号外显子C797S点突变是介导第三代EGFR-TKIs耐药的最常见的机制之一。C797S是丝氨酸取代了半胱氨酸的错义突变,位于EGFR的酪氨酸激酶区,使第三代EGFR-TKI无法在ATP结合域内继续形成共价键,导致EGFR-TKI失去了阻断EGFR通路的作用。AURA3研究中73例二线使用奥西替尼后进展的患者血浆循环肿瘤DNA(circulating tumour DNA, ctDNA)基因组测序结果显示,有10例(14%)患者发生了C797S突变,且均保留了T790M突变,C797S突变与T790M突变为顺式[10]。Le等在42例奥西替尼耐药的患者中发现了8例(19%)C797S突变,且仅在T790M突变保留的病例中出现[12]。Oxnard和Lin等的研究也提示C797S突变似乎只发生在疾病进展后维持T790M突变的细胞中[13-14]。在FLAURA研究中,奥西替尼耐药后出现C797S突变的频率为7%(6/91)[11]。可见奥西替尼作为一线和二线治疗NSCLC的耐药机制存在差异。除奥西替尼外,还有研究报道了C797S突变可介导对其他第三代TKI的耐药,如Olmutinib、Rociletinib和Nazartinib[15]。
AURA3和FLAURA研究中观察到的其他与耐药相关的罕见EGFR点突变还有L792X(3%)、G796S(1%)和L718Q(1%)突变、20外显子插入(1%)、L7128Q(2%)、S768I(1%)[10-11]。奥西替尼治疗对G719S、G724S等罕见突变可能无效,L792和L718突变可显著增加奥西替尼的半抑制浓度,其中L718Q突变对耐药性的影响最大,进一步的研究显示L718Q突变对所有第三代TKI表现出交叉耐药性[16]。表明这些点突变可以引起空间干扰,导致化合物对EGFR激酶结构域的亲和力降低。
2.1.2 T790M缺失入选AURA3研究的患者均在一线使用一、二代EGFR-TKIs治疗后出现了T790M突变,其中49%(36/73)在奥西替尼耐药后出现了T790M缺失[10]。Lin等发现奥西替尼耐药的18例患者中8例(44%)失去了T790M突变,但均维持了EGFR激活突变[14]。Le[12]和Oxnard[13]等的研究中耐药患者T790M缺失的概率分别为53%(21/40)和68%(28/41)。Oxnard等通过对奥西替尼耐药后110例患者ctDNA检测结果进行分析,提出耐药时T790M的缺失与1~3周治疗后EGFR驱动基因突变水平小幅下降有关[13]。Piotrowska等[17]报道的Rociletinib耐药的患者中也有50%(6/12)出现T790M缺失。最近一项研究发现奥西替尼耐药后T790M缺失患者较保留T790M突变的患者mPFS(4.4月vs. 7.7月)更短[18]。而在FLAURA研究中一线使用奥西替尼治疗的患者耐药后未出现T790M突变[11]。
T790M缺失可能是第三代EGFR-TKI治疗后的结果,也可能是导致耐药的原因之一,与肿瘤进展过程中肿瘤的异质性有关。在T790M保留的病例中,耐药机制大部分与C797S突变或旁路异常激活有关,而T790M缺失的患者常表现出不同的耐药机制,大多不依赖EGFR信号通路[12]。在治疗过程中重复进行T790M突变状态检测有助于对第三代EGFR-TKIs耐药机制和后续治疗策略的研究。
2.1.3 EGFR扩增Nukaga等[19]在Rociletinib耐药的PC-9细胞系中发现了扩增的EGFR基因。Piotrowska等[17]报道了25%(3/12)Rociletinib治疗耐药的患者出现EGFR的扩增。1例奥西替尼耐药的患者在治疗过程中出现了EGFR扩增,扩增水平与影像学进展相平行[20]。Le等研究发现42例奥西替尼耐药的患者中有8名(19%)发生了EGFR扩增[12]。可见,EGFR基因扩增更多见于Rociletinib治疗后的患者,耐药机制可能是由于EGFR基因扩增导致TKIs类药物浓度相对不足。
2.2 旁路异常激活 2.2.1 MET和HER2扩增间质表皮转化因子(mesenchymal-epithelial transition factor, MET)和人表皮生长因子受体2(human epidermal growth factor receptor 2, HER2)的扩增在第一、三代EGFR-TKIs的耐药机制中均占有重要地位。AURA3研究结果显示奥西替尼作为二线治疗耐药后有19%(14/73)的患者出现了MET扩增,是C797S突变后第二常见的耐药机制,5%(4/73)的患者出现了HER2扩增,一例患者同时发生了MET和HER2扩增[10]。Le等[12]研究中MET扩增的发生率为14%(6/42),HER2扩增为10%(4/42)。Lin等[14]研究发现有MET扩增和无MET扩增的患者在mPFS、中位总生存期(median overall survival, mOS)和进展后存活率上,差异均无统计学意义。FLAURA研究中,奥西替尼作为一线治疗,耐药后的患者有15%(14/91)出现了MET扩增,超过了C797S突变所占比例7%(6/91),HER2扩增的发生率为2%(2/91)[11]。可见,当奥西替尼用于一线治疗时,MET扩增将成为最常见的耐药机制。
2.2.2 FGFR信号通路异常成纤维细胞生长因子受体(fibroblast growth factor receptor, FGFR)是一类跨膜的酪氨酸激酶受体。Kim等[21]发现在奥西替尼耐药的患者中出现了FGFR1扩增和成纤维细胞生长因子2(fibroblast growth factor 2, FGF2)mRNA增加,表明FGF2-FGFR1自分泌环可能与耐药有关。两例T790M突变的患者在接受奥西替尼和Naquotinib治疗后进展,ctDNA检测均发现了FGFR3-TACC3融合突变。AURA3研究中奥西替尼耐药的患者出现FGFR3-TACC3融合的概率为1%(1/73)[10]。提示FGFR信号通路异常可能是第三代EGFR-TKIs的获得性耐药机制之一。
2.2.3 IGF1R异常激活Park等[22]在WZ4002的耐药细胞株中发现了胰岛素样生长因子1受体(insulin-like growth factor 1 receptor, IGF1R)的异常激活及胰岛素样生长因子结合蛋白3(insulin-like growth factor-binding protein 3, IGFBP3)的缺失,使用shRNA下调IGF1R或使用IGF1R抑制剂均会使耐药细胞株恢复对WZ4002的敏感性。表明IGF1R异常活化可能是第三代EGFR-TKI的耐药机制之一。
2.3 下游信号通路异常激活 2.3.1 RAS-MAPK通路活化第三代EGFR-TKIs继发性耐药与RAS基因-有丝分裂原活化蛋白激酶(mitogen-activated protein kinase, MAPK)通路激活有关。Nakatani等[23]在奥西替尼耐药的细胞系中观察到了KRAS基因扩增和KRAS蛋白过表达。AURA3研究中有1例(1%)患者耐药后出现了KRAS(G12D)基因突变,2例(3%)出现BRAF V600E突变[10]。Oxnar等[13]在奥西替尼耐药的患者中检测到了1例(2%)KRAS(Q61K)基因突变、2例(5%)BRAF突变和1例(2%)ESYT2-BRAF基因融合。FLAURA研究中一线使用奥西替尼治疗的患者耐药后发生的KRAS突变包括:A146T(1%)、G12C(1%)、G12D(1%),BRAF V600E突变发生率为3%(3/91)[11]。一例奥西替尼一线治疗后进展的患者同时出现了BRAF V600E突变和MET扩增。
2.3.2 PI3K/AKT/mTOR通路活化1-磷酸酰肌醇-3-激酶(phosphatidylinositol 3-kinases, PIK3CA)是肺腺癌的致癌驱动基因之一,其突变可以促进肿瘤的浸润、提高其下游激酶PI3Ks的活性。AURA3研究中,奥西替尼耐药的患者PIK3CA扩增的发生率为4%(3/73),1%(1/73)的患者出现了PIK3CA E545K突变[10]。Oxnard等[13]报道的耐药患者中PIK3CA扩增的发生率为10%(4/41)。FLAURA研究中一线使用奥西替尼治疗的患者耐药后发生的PIK3CA突变包括:E453K(1%)、E545K(4%)、H1047R(1%)[11]。由此推测PIK3CA突变或扩增使PI3Ks活性增加,并引起各种下游激酶激活,导致PI3K/AKT/mTOR通路活化,从而与上游EGFR磷酸化解偶联,可能是第三代EGFR-TKI的耐药机制之一。
2.4 组织学转化Marcoux等[24]报道了19例NSCLC患者在第三代EGFR-TKIs治疗后发生小细胞肺癌(small cell lung cancer, SCLC)转化,以Rb1、TP53和PIK3CA突变为特征,中位转化时间为17.8月,转化后的mOS为10.9月。Lin等[14]对40例奥西替尼耐药的患者进行ctDNA检测,其中2例发生了SCLC转化,1例出现了鳞状细胞癌(squamous cell carcinoma, SCC)转化。有研究汇总分析了至今为止报道的16例第三代TKIs治疗后发生SCC转化的病例,发现82%的患者为女性,中位转化时间为11.5月,均维持基础EGFR突变,无论接受何种治疗,转化后的mOS为3.5月[25]。目前尚未在体外研究中发现TKIs耐药后SCC转化的现象,说明肿瘤微环境在这一转化过程中可能起着潜在作用。Nukaga等[19]通过体外研究发现对奥西替尼和Rociletinib耐药的H1975细胞表现出梭形细胞样形态并且获得了更多的间充质表型,证实了上皮—间质细胞转化(epithelial-mesenchymal transition, EMT)与第三代EGFR-TKI耐药密切相关。由此可见,对于第三代EGFR-TKIs耐药的患者,除了液体活检明确相关突变基因之外,通过组织活检来明确有无组织学类型的转变也十分重要,但迄今为止尚未在临床上报道关于EMT与第三代EGFR-TKI耐药的明确证据。
2.5 其他 2.5.1 细胞周期基因改变AURA3和FLAURA研究发现,奥西替尼作为一线或二线治疗,进展后的患者出现了细胞周期基因改变的频率分别为11%和12%[10-11]。在AURA3研究中CCNE1扩增和CDK6扩增的出现频率均为7%(5/73),为最常见的细胞周期基因改变[10]。FLAURA研究中最常见的细胞周期基因改变为CDK6扩增(3%)[11]。在Le等的研究中还发现了CCND1、CCNE1、CDKN2A缺失[12]。细胞周期基因改变可能参与了奥西替尼耐药的发生。
2.5.2 致癌基因融合AURA3研究奥西替尼耐药的患者中发现1例RET-ERC1和1例NTRK1-TPM3基因融合[10]。Oxnar等[13]报道了1例奥西替尼耐药后出现CCDC6-RET基因融合。FLAURA研究中发现1例ALK基因融合[11]。Schrock等[26]收集了31例EGFR-TKIs耐药后出现致癌基因融合的患者,包括BRAF、ALK、RET、FGFR3等,其中一例T790M突变的患者在奥西替尼治疗进展后,ctDNA检测发现PLEKHA7-ALK基因融合,联合使用奥西替尼和ALK抑制剂Alectinib治疗后,患者病情缓解,4月后重复进行ctDNA检测发现PLEKHA7-ALK基因融合消失。提示致癌基因融合可能是EGFR-TKIs的潜在耐药机制。
2.5.3 氧化供能途径的改变Martin等[27]研究发现,奥西替尼抑制肿瘤细胞的作用与其抑制糖酵解有关。对奥西替尼耐药的细胞可以调节其能量源从葡萄糖到乳糖或半乳糖,通过氧化磷酸化(oxidative phosphorylation, OxPhos)来获取ATP,而不依赖于糖酵解和EGFR通路,导致其对奥西替尼的敏感性下降。
3 应对策略 3.1 EGFR-TKIs治疗C797S突变C797S突变所在染色体的基因状态会影响后续治疗,临床前研究为制定后续治疗策略提供了指导。针对第三代EGFR-TKIs的不同耐药机制的应对策略,见图 3。

|
| 图 3 基于体外研究得到的针对第三代EGFR-TKIs治疗晚期EGFR突变NSCLC不同耐药机制的应对策略 Figure 3 Therapeutic perspectives identified according to in vitro studies for resistance mechanisms of advanced EGFR-mutated NSCLC to the third-generation EGFR-TKIs |
若C797S和T790M突变为顺式结构(位于同一等位基因)(85%),则对第一到三代EGFR-TKIs均耐药[28]。第四代EGFR-TKI EAI045是第一个针对EGFR中T790M和C797S突变的变构TKI。体外研究发现,EAI045联合西妥昔单抗能显著抑制具有EGFR L858R/T790M/C797S突变的细胞系增殖,在小鼠移植瘤模型中也有效,单用则疗效不佳,且对EGFR del19/C797S/T790M突变导致的耐药无效[29]。EAI045和西妥昔单抗联合作用于L792H或G796R与T790M顺式结合的细胞系效力很低,但多西紫杉醇却能有效抑制突变细胞增殖。德国科学家Gunther等开发了一类新的三取代咪唑类化合物,它们对L858R/T790M/C797S突变细胞的选择敏感性较野生型更高[15]。Brigatinib是ALK和EGFR抑制剂,对del19/C797S/T790M和L858R/C797S/T790M突变细胞系均有效,但对del19/C797S/T790M的敏感性更强,其抑制作用在小鼠模型中也得到了证实,联合西妥昔单抗或帕尼单抗能提高其疗效[30]。还需要临床试验来验证新的EGFR-TKIs在晚期NSCLC患者中的疗效。
3.1.2 第三代TKI联合第一代TKI当C797S和T790M突变为反式结构(位于不同等位基因)(10%),肿瘤对第三代TKIs耐药,但对第一代和第三代TKIs联合治疗敏感[28]。一例患者在奥西替尼治疗后发生T790M和C797S反式突变并耐药,厄洛替尼和奥西替尼联合治疗有效,但3月后再次出现耐药,且重复液体活检显示T790M和C797S突变为顺式。
3.1.3 第一、二代EGFR-TKIs体外研究发现,具有del19/C797S突变,且不伴有T790M突变的细胞系对第三代TKIs具有耐药性,但对吉非替尼或阿法替尼保持敏感[28]。一例患者在奥西替尼治疗后进展并出现del19/C797S突变,在使用吉非替尼治疗后肿瘤部分缓解。表明一线接受第三代TKIs治疗的T790M阴性患者,在C797S的驱动下获得耐药性,且不伴有其他替代竞争耐药机制,则后续治疗可能对第一、二代TKIs敏感。
3.2 第三代TKIs联合其他通路抑制剂对于其他耐药机制引起的耐药,第三代TKIs联合针对耐药机制的通路抑制剂是后续的治疗策略。目前正在进行的第三代EGFR-TKIs联合靶向及免疫治疗的临床研究,见表 1。
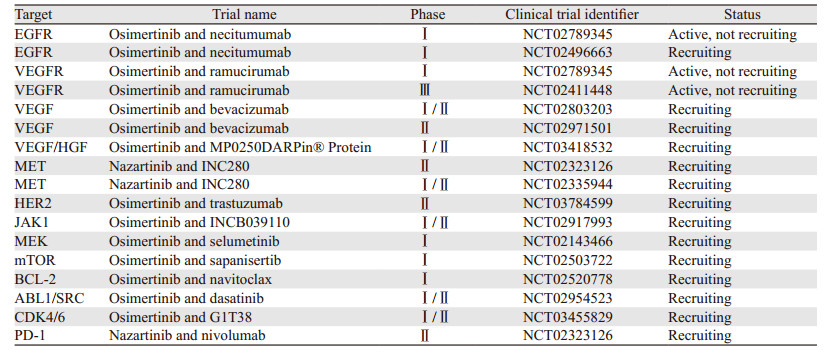
|
针对MET或HER2扩增导致的耐药,MET或HER2抑制剂单用或与第三代EGFR-TKIs联合是潜在的治疗策略。临床前研究发现MET高表达的HCC827细胞株对奥西替尼及Rociletinib均耐药,加用MET抑制剂后对奥西替尼的敏感性升高[31]。Monica等联合使用奥西替尼和HER2抑制剂曲妥珠单抗克服了PC9细胞对奥西替尼的耐药性,并且延缓了H1975细胞对奥西替尼耐药性的产生,在小鼠移植瘤模型中也得到了相同的结果[32]。1例奥西替尼耐药的患者出现高水平的MET扩增,给予MET抑制剂克唑替尼单药治疗后呼吸困难等症状缓解,病灶维持稳定。
3.2.2 MEK抑制剂对于RAS-MAPK信号通路激活引起的耐药,第三代EGFR-TKIs联合丝裂原活化的细胞外信号调节激酶(mitogen-activated extracellular signal-regulated kinase, MEK)抑制剂是潜在的治疗策略。临床前研究发现,奥西替尼联合Selumetinib可以克服KRAS扩增和过表达引起的耐药[23],联合Trametinib可以克服KRAS突变引起的耐药。另一项体外研究中,MEK抑制剂通过抑制BIM和髓样细胞白血病-1(myeloid cell leukemia-1, MCL-1)的细胞外信号调节激酶(extracellular signal-regulated kinase, ERK)依赖性磷酸化恢复了del19/T790M/C797S突变的PC9细胞系对奥西替尼的敏感性,提示靶向MEK/ERK信号转导是克服del19/T790M/C797S突变的另一种可能的策略[33]。
3.2.3 VEGFR抑制剂血管内皮生长因子(vascular endothelial growth factor, VEGF)和EGFR通路密切相关。1例晚期肺腺癌患者通过联合使用奥西替尼和贝伐珠单抗治疗有效,但3月后疾病进展,加用Brigatinib三药联合治疗1月后病灶部分缓解。另外1例晚期肺腺癌患者二线使用奥西替尼治疗8月后进展,但在阿帕替尼治疗3周后原发肿瘤和转移灶均缩小[34]。奥西替尼联合雷莫芦单抗或贝伐珠单抗治疗EGFR突变的晚期NSCLC患者的Ⅰ/Ⅱ期临床试验正在进行中。
3.2.4 OxPhos抑制剂第三代TKI联合OxPhos抑制剂可能克服氧化供能途径的改变引起的耐药。体外研究发现,OxPhos抑制剂Phenformin和奥西替尼联合可增加细胞对奥西替尼的敏感性,并延缓奥西替尼耐药的出现。而在动物实验中也证实联合用药能推迟肿瘤复发的时间[27]。
3.2.5 新靶点抑制剂Takahashi等[35]发现锚蛋白重复结构域1(ankyrin repeat domain 1, ANKRD1)与EMT有关,且在第三代EGFR-TKIs耐药细胞中过表达,伊马替尼可抑制ANKRD1的表达使耐药细胞恢复对奥西替尼的敏感性。Yochum等[36]报道了转录因子TWIST1抑制剂Harmine和Bcl-2/BclXL抑制剂ABT737能够靶向EGFR突变NSCLC中的EMT转录因子TWIST1,它们和奥西替尼联合使用时能抑制TWIST1过表达,同时使BIM表达增加,从而克服EMT和BIM介导的耐药性。Tanimoto等[7]体外研究发现,组蛋白去乙酰化酶(histone deacetylase, HDAC)抑制剂可以上调EGFR突变NSCLC细胞中BIM的表达从而克服耐药,联合应用奥西替尼和HDAC抑制剂伏立诺他可引起存在BIM缺失多态性的耐药细胞凋亡。
3.3 其他策略 3.3.1 针对EGFR 20外显子插入突变Poziotinib是一种不可逆的HER2抑制剂,在体外研究中能有效抑制EGFR 20外显子插入突变的细胞系,有效性是奥西替尼的100倍。在Ⅱ期临床研究中,Poziotinib治疗EGFR 20外显子插入突变NSCLC患者的客观缓解率(objective response rates, ORR)为64%,mOS仍未达到[9]。Luminespib是Hsp90抑制剂,在体外研究中能降解EGFR 20外显子插入突变并诱导细胞凋亡。1例EGFR 20外显子插入突变患者在luminespib治疗后部分缓解,其Ⅱ期临床研究结果显示ORR为17%,11/29(38%)的患者PR或SD≥3月[37]。Poziotinib和Luminespib为EGFR 20外显子插入突变导致原发性耐药的NSCLC的治疗带来了希望。
3.3.2 免疫治疗免疫治疗或免疫联合第三代TKIs能否成为患者对第三代TKIs耐药后的治疗策略仍需进一步研究。回顾性分析显示,抗PD-1单药治疗EGFR突变阳性患者疗效差,即使对于PD-L1表达≥50%的患者也是如此[38]。奥西替尼联合Durvalumab在EGFR T790M突变患者中的客观缓解率为67%,但间质性肺病的发生率为38%[39],该研究已因不良反应而暂停。免疫治疗联合化疗或许能为第三代TKIs耐药的患者带来新希望。Oh等[17]报道了1例耐药后出现C797S突变的患者经过吉西他滨和顺铂化疗两周期、帕博利珠单抗治疗6周期后重复活检提示C797S突变消失,此后再次使用奥西替尼反应良好。Nivolumab联合化疗用于对EGFR-TKI耐药的EGFR突变/T790M阴性NSCLC患者以及帕博利珠单抗联合化疗治疗奥西替尼一线治疗失败的NSCLC患者的临床试验(NCT03515837)目前正在进行中。
3.3.3 化疗若患者的身体状态允许,耐药后再行化疗也是一种选择,尤其是对于发生了SCLC转化的患者,但目前尚缺乏充分研究。Oh等[18]报道了2例奥西替尼治疗后进展的患者发生了SCLC转化,对依托泊苷和顺铂产生了良好的反应。Marcoux等[24]也发现发生SCLC转化后的患者经依托泊苷、顺铂和紫杉醇治疗后均产生了较高的临床缓解率。而对于耐药后没有可靶向的突变或耐药机制不详的患者,可以选择顺铂/卡铂加紫杉醇/白蛋白紫杉醇/培美曲塞,加或不加贝伐珠单抗,持续4到6周期后,再使用培美曲塞、多西他赛或厄洛替尼单药维持治疗。
4 小结第三代EGFR-TKIs治疗晚期NSCLC的耐药机制十分复杂,它作为一线和二线治疗的耐药机制有所不同,且EGFR敏感突变的肿瘤对第三代TKIs产生耐药的机制在不同患者之间和同一患者肿瘤的不同部位都存在异质性,可能仍有一些机制目前尚未明确。因此,ctDNA动态检测或组织活检明确耐药机制对于指导下一步的治疗十分重要,由于肿瘤的异质性,ctDNA的检测可能会比组织活检更能反映疾病的全貌。只有从机制入手,研究针对耐药靶点的新靶向药物或新的联合治疗方案,才能为耐药患者制定出更合理的治疗策略。
作者贡献
何婧怡:文献搜集与整理,综述撰写与修改
吴芳:文献提供,指导论文修改
胡春宏:文章框架构思与制定,指导论文修改
| [1] | Wu YL, Zhong WZ, Li LY, et al. Epidermal growth factor receptor mutations and their correlation with gefitinib therapy in patients with non-small cell lung cancer: a meta-analysis based on updated individual patient data from six medical centers in mainland China[J]. J Thorac Oncol, 2007, 2(5): 430–439. DOI:10.1097/01.JTO.0000268677.87496.4c |
| [2] | Siegel RL, Miller KD, Jemal A. Cancer statistics, 2016[J]. CA Cancer J Clin, 2016, 66(1): 7–30. DOI:10.3322/caac.21332 |
| [3] | Mok TS, Wu Y-L, Ahn M-J, et al. Osimertinib or platinum-pemetrexed in EGFR T790M-positive lung cancer[J]. N Engl J Med, 2017, 376(7): 629–640. DOI:10.1056/NEJMoa1612674 |
| [4] | Soria JC, Ohe Y, Vansteenkiste J, et al. Osimertinib in untreated EGFR-mutated advanced non-small-cell lung cancer[J]. N Engl J Med, 2018, 378(2): 113–125. DOI:10.1056/NEJMoa1713137 |
| [5] | Li X, Wang S, Li B, et al. BIM Deletion Polymorphism Confers Resistance to Osimertinib in EGFR T790M Lung Cancer: a Case Report and Literature Review[J]. Target Oncol, 2018, 13(4): 517–523. DOI:10.1007/s11523-018-0573-2 |
| [6] | Zhao M, Zhang Y, Cai W, et al. The Bim deletion polymorphism clinical profile and its relation with tyrosine kinase inhibitor resistance in Chinese patients with non-small cell lung cancer[J]. Cancer, 2014, 120(15): 2299–2307. DOI:10.1002/cncr.28725 |
| [7] | Tanimoto A, Takeuchi S, Arai S, et al. Histone Deacetylase 3 Inhibition Overcomes BIM Deletion Polymorphism-Mediated Osimertinib Resistance in EGFR-Mutant Lung Cancer[J]. Clin Cancer Res, 2017, 23(12): 3139–3149. DOI:10.1158/1078-0432.CCR-16-2271 |
| [8] | Arcila ME, Nafa K, Chaft JE, et al. EGFR exon 20 insertion mutations in lung adenocarcinomas: prevalence, molecular heterogeneity, and clinicopathologic characteristics[J]. Mol Cancer Ther, 2013, 12(2): 220–229. |
| [9] | Robichaux JP, Elamin YY, Tan Z, et al. Mechanisms and clinical activity of an EGFR and HER2 exon 20-selective kinase inhibitor in non-small cell lung cancer[J]. Nat Med, 2018, 24(5): 638–646. DOI:10.1038/s41591-018-0007-9 |
| [10] | Papadimitrakopoulou VA, Wu YL, Han JY, et al. Analysis of resistance mechanisms to osimertinib in patients with EGFR T790M advanced NSCLC from the AURA3 study[J]. Ann Oncol, 2018, 29(Suppl 8): LBA51. |
| [11] | Ramalingam SS, Cheng Y, Zhou C, et al. Mechanisms of acquired resistance to first-line osimertinib: Preliminary data from the phase Ⅲ FLAURA study[J]. Ann Oncol, 2018, 29(Suppl 8): LBA50. |
| [12] | Le X, Puri S, Negrao MV, et al. Landscape of EGFR-dependent and-independent resistance mechanisms to osimertinib and continuation therapy beyond progression in EGFR-mutant NSCLC[J]. Clin Cancer Res, 2018, 24(24): 6195–6203. DOI:10.1158/1078-0432.CCR-18-1542 |
| [13] | Oxnard GR, Hu Y, Mileham KF, et al. Assessment of Resistance Mechanisms and Clinical Implications in Patients With EGFR T790M-Positive Lung Cancer and Acquired Resistance to Osimertinib[J]. JAMA Oncol, 2018, 4(11): 1527–1534. DOI:10.1001/jamaoncol.2018.2969 |
| [14] | Lin CC, Shih JY, Yu CJ, et al. Outcomes in patients with non-small-cell lung cancer and acquired Thr790Met mutation treated with osimertinib: a genomic study[J]. Lancet Respir Med, 2018, 6(2): 107–116. DOI:10.1016/S2213-2600(17)30480-0 |
| [15] | Lu X, Yu L, Zhang Z, et al. Targeting EGFRL858R/T790M and EGFRL858R/T790M/C797S resistance mutations in NSCLC: Current developments in medicinal chemistry[J]. Med Res Rev, 2018, 38(5): 1550–1581. DOI:10.1002/med.21488 |
| [16] | Bersanelli M, Minari R, Bordi P, et al. L718Q mutation as new mechanism of acquired resistance to AZD9291 in EGFR-mutated NSCLC[J]. J Thorac Oncol, 2016, 11(10): e121–123. DOI:10.1016/j.jtho.2016.05.019 |
| [17] | Piotrowska Z, Niederst MJ, Karlovich CA, et al. Heterogeneity underlies the emergence of EGFRT790 wild-type clones following treatment of T790M-positive cancers with a third-generation EGFR inhibitor[J]. Cancer Discov, 2015, 5(7): 713–722. DOI:10.1158/2159-8290.CD-15-0399 |
| [18] | Oh DK, Ji WJ, Kim WS, et al. Efficacy, safety, and resistance profile of osimertinib in T790M mutation-positive non-small cell lung cancer in real-world practice[J]. PLoS One, 2019, 14(1): e0210225. DOI:10.1371/journal.pone.0210225 |
| [19] | Nukaga S, Yasuda H, Tsuchihara K, et al. Amplification of EGFR wild-type alleles in non-small cell lung cancer cells confers acquired resistance to mutation-selective EGFR tyrosine kinase inhibitors[J]. Cancer Res, 2017, 77(8): 2078–2089. DOI:10.1158/0008-5472.CAN-16-2359 |
| [20] | Knebel FH, Bettoni F, Shimada AK, et al. Sequential liquid biopsies reveal dynamic alterations of EGFR driver mutations and indicate EGFR amplification as a new mechanism of resistance to osimertinib in NSCLC[J]. Lung Cancer, 2017, 108: 238–241. DOI:10.1016/j.lungcan.2017.04.004 |
| [21] | Kim TM, Song A, Kim DW, et al. Mechanisms of acquired resistance to AZD9291: A mutation-selective, irreversible EGFR inhibitor[J]. J Thorac Oncol, 2015, 10(12): 1736–1744. DOI:10.1097/JTO.0000000000000688 |
| [22] | Park JH, Choi YJ, Kim SY, et al. Activation of the IGF1R pathway potentially mediates acquired resistance to mutant-selective 3rd-generation EGF receptor tyrosine kinase inhibitors in advanced non-small cell lung cancer[J]. Oncotarget, 2016, 7(16): 22005–22015. |
| [23] | Nakatani K, Yamaoka T, Ohba M, et al. KRAS and EGFR Amplifications Mediate Resistance to Rociletinib and Osimertinib in Acquired Afatinib-Resistant NSCLC Harboring Exon 19 Deletion/T790M in EGFR[J]. Mol Cancer Ther, 2019, 18(1): 112–126. |
| [24] | Marcoux N, Gettinger SN, O'Kane G, et al. EGFR-mutant adenocarcinomas that transform to small-cell lung cancer and other neuroendocrine carcinomas: clinical outcomes[J]. J Clin Oncol, 2019, 37(4): 278–285. DOI:10.1200/JCO.18.01585 |
| [25] | Roca E, Pozzari M, Vermi W, et al. Outcome of EGFR-mutated adenocarcinoma NSCLC patients with changed phenotype to squamous cell carcinoma after tyrosine kinase inhibitors: A pooled analysis with an additional case[J]. Lung Cancer, 2019, 127: 12–18. DOI:10.1016/j.lungcan.2018.11.016 |
| [26] | Schrock AB, Zhu VW, Hsieh WS, et al. Receptor Tyrosine Kinase Fusions and BRAF Kinase Fusions are Rare but Actionable Resistance Mechanisms to EGFR Tyrosine Kinase Inhibitors[J]. J Thorac Oncol, 2018, 13(9): 1312–1323. DOI:10.1016/j.jtho.2018.05.027 |
| [27] | Martin MJ, Eberlein C, Taylor M, et al. Inhibition of oxidative phosphorylation suppresses the development of osimertinib resistance in a preclinical model of EGFR-driven lung adenocarcinoma[J]. Oncotarget, 2016, 7(52): 86313–86325. |
| [28] | Niederst MJ, Hu H, Mulvey HE, et al. The allelic context of the C797S mutation acquired upon treatment with third-generation EGFR inhibitors impacts sensitivity to subsequent treatment strategies[J]. Clin Cancer Res, 2015, 21(17): 3924–3933. DOI:10.1158/1078-0432.CCR-15-0560 |
| [29] | Jia Y, Yun CH, Park E, et al. Overcoming EGFR (T790M) and EGFR (C797S) resistance with mutant-selective allosteric inhibitors[J]. Nature, 2016, 534(7605): 129–132. DOI:10.1038/nature17960 |
| [30] | Uchibori K, Inase N, Araki M, et al. Brigatinib combined with anti-EGFR antibody overcomes osimertinib resistance in EGFR-mutated non-small cell lung cancer[J]. Nat Commun, 2017, 8: 14768. DOI:10.1038/ncomms14768 |
| [31] | Shi P, Oh YT, Zhang G, et al. Met gene amplification and protein hyperactivation is a mechanism of resistance to both first and third generation EGFR inhibitors in lung cancer treatment[J]. Cancer Lett, 2016, 380(2): 494–504. DOI:10.1016/j.canlet.2016.07.021 |
| [32] | La Monica S, Cretella D, Bonelli M, et al. Trastuzumab emtansine delays and overcomes resistance to the third-generation EGFR-TKI osimertinib in NSCLC EGFR mutated cell lines[J]. J Exp Clin Cancer Res, 2017, 36(1): 174. DOI:10.1186/s13046-017-0653-7 |
| [33] | Shi P, Oh YT, Deng L, et al. Overcoming acquired resistance to AZD9291, a third-generation EGFR inhibitor, through modulation of ERK/ERK-dependent Bim and Mcl-1 degradation[J]. Clin Cancer Res, 2017, 23(21): 6567–6579. DOI:10.1158/1078-0432.CCR-17-1574 |
| [34] | Yang D, Dai R, Zhang Q, et al. Apatinib for heavily treated patients with non-small cell lung cancer: Report of a case series and literature review[J]. Saudi J Biol Sci, 2018, 25(5): 888–894. DOI:10.1016/j.sjbs.2017.12.011 |
| [35] | Takahashi A, Seike M, Chiba M, et al. Ankyrin Repeat Domain 1 Overexpression is Associated with Common Resistance to Afatinib and Osimertinib in EGFR-mutant Lung Cancer[J]. Sci Rep, 2018, 8(1): 14896. DOI:10.1038/s41598-018-33190-8 |
| [36] | Yochum ZA, Cades J, Wang H, et al. Targeting the EMT transcription factor TWIST1 overcomes resistance to EGFR inhibitors in EGFR-mutant non-small-cell lung cancer[J]. Oncogene, 2019, 38(5): 656–670. DOI:10.1038/s41388-018-0482-y |
| [37] | Piotrowska Z, Costa DB, Oxnard GR, et al. Activity of the Hsp90 inhibitor luminespib among non-small-cell lung cancers harboring EGFR exon 20 insertions[J]. Ann Oncol, 2018, 29(10): 2092–2097. |
| [38] | Lisberg A, Cummings A, Goldman JW, et al. A Phase Ⅱ Study of Pembrolizumab in EGFR-Mutant, PD-L1+, Tyrosine Kinase Inhibitor Naïve Patients With Advanced NSCLC[J]. J Thorac Oncol, 2018, 13(8): 1138–1145. DOI:10.1016/j.jtho.2018.03.035 |
| [39] | Ahn MJ, Yang J, Yu H, et al. Osimertinib combined with durvalumab in EGFR-mutant non-small cell lung cancer: results from the TATTON phase Ib trial[J]. J Thorac Oncol, 2016, 11(Suppl 4): S115. |