 2019, Vol. 46
2019, Vol. 46文章信息
- 小细胞肺癌关键基因及信号通路分析
- Key Genes and Signaling Pathways in Small Cell Lung Cancer
- 肿瘤防治研究, 2019, 46(8): 683-689
- Cancer Research on Prevention and Treatment, 2019, 46(8): 683-689
- http://www.zlfzyj.com/CN/10.3971/j.issn.1000-8578.2019.18.2039
- 收稿日期: 2019-01-02
- 修回日期: 2019-04-28
引用本文 |



2. 100029 北京,北京中医药大学研究生院
2. Graduate School, Beijing University of Chinese Medicine, Beijing 100029, China
肺癌是世界范围内发病率和死亡率最高的恶性肿瘤之一[1],其中小细胞肺癌(small cell lung cancer, SCLC)作为神经内分泌肿瘤,约占全部肺癌的15%,5年生存率 < 6%,具有侵袭性强、易早期血行和淋巴转移、病死率高等特点[2]。常规的治疗方式包括化疗、放疗和手术等,化疗是最主要的治疗方式,但存在耐药性、易复发等问题。在过去的几十年中,SCLC患者的生存率没有得到明显改善,并且暂无分子靶向药物被证明能够明显延长患者生存期[3]。SCLC相关基因及信号通路的改变促进细胞增殖和抑制细胞凋亡,导致肿瘤细胞的早期转移,如PI3K/AKT/mTOR通路中的PIK3CA、PTEN、Akt、RICTOR等基因的突变、甲基化或表达水平的变化[4],因此亟需进一步探讨SCLC分子机制,以期寻找SCLC早期诊断及靶向治疗潜在的生物标志物。近年来,基因芯片技术和生物信息学分析广泛应用于基因组学的研究,但由于SCLC侵袭性强、生存期短等特点导致相关基因芯片数据较少,如Rohrbeck等[5]分析肺癌mRNA的表达情况,发现CDK、NCAM1、DEK等基因的异常表达与SCLC的发生具有相关性。本研究整合公共基因芯片数据库(Gene Expression Omnibus, GEO)中多个SCLC基因表达的芯片数据集,通过生物信息学技术寻找差异表达基因(differentially expressed genes, DEGs),进行功能分析并构建蛋白互作网络,从而更好地了解SCLC的分子机制。
1 材料与方法 1.1 小细胞肺癌mRNA数据集筛选利用NCBI(National Center for Biotechnology Information)平台的GEO数据库(http://www.ncbi.nlm.nih.gov/geo/)进行基因芯片筛选,GEO是一个公开的基因组数据库,包含了整个基因表达数据、芯片和微阵列。目标芯片的准入标准:(1)临床患者SCLC标本,排除细胞株和动物实验;(2)入选芯片需含有SCLC癌组织标本和正常组织标本;(3)仅为mRNA芯片,且已进行标准化。
1.2 差异表达基因筛选根据“1.1”纳入标准,确定目标芯片后,利用在线工具GEO2R分析各个芯片数据,设置筛选标准为:错误发现率(false discovery rate, FDR) < 0.05,|log FC| > 1(fold change, FC),并将目标基因探针名称转化为标准基因名称,对所筛选出的芯片进行数据下载,并筛选出DEGs。
1.3 基因功能注释与通路富集分析DAVID(Database for Annotation, Visualization and Integrated Discovery Database)生物信息资源数据库整合了生物数据和分析工具,能够对基因和蛋白质进行功能注释。通过DAVID进行在线分析,以人源基因为背景进行基因本体(Gene Ontology, GO)和基因组百科全书数据库(Kyoto Encyclopedia of Genes and Genomes, KEGG)对DEGs进行富集分析,筛选出SCLC组织与正常组织之间有生物特征差异的基因簇和通路,并使用Cytoscape3.6.0软件的插件BinGO绘制GO富集可视化互交网络。
1.4 蛋白互作网络分析Cytoscape3.6.0作为生物信息学软件平台,通过构建蛋白互作网络将分子交互网络可视化;本研究通过Cytoscape3.6.0软件构建DEGs的蛋白互作网络以发现SCLC的关键基因。MCODE(Molecular Complex Detection Technology)是Cytoscape3.6.0软件中对构建的生物学网络进行关联度分析的插件,根据关联积分值,可获得整个网络中可能形成的蛋白质簇和关键节点蛋白,并在Cytoscape3.6.0软件中进行可视化显示。通过MCODE插件筛选核心基因的标准如下:degree cutoff=2,node score cutoff=0.2,k-core=2和max.depth=100。
1.5 核心基因与预后的关系通过GEO数据库的挖掘,明确差异表达的核心基因,利用Kaplan-Meier在线网站(http://kmplot.com/analysis/)分析核心基因表达水平与预后的关系,并绘制生存曲线。
2 结果 2.1 差异表达基因根据纳入标准的要求,共筛选出2个符合要求的mRNA微阵列数据集,分别为GSE6044[5]和GSE40275(https://www.ncbi.nlm.nih.gov/geo/query/acc.cgi)。GSE6044包含9例SCLC组织样本和5例正常肺组织样本,GSE40275包含8例SCLC组织样本和14例正常肺组织样本。17例SCLC者(男11例,女6例)年龄39~80岁,平均年龄(65.37±10.14)岁;19例正常肺组织样本提供者(男9例,女10例)年龄38~80岁,平均年龄(67.69±7.23)岁。GEO2R分析发现2个数据集共表达的差异基因有248个,其中172个为高表达基因和76个低表达基因,本文列举|log FC|最大的20个基因,见表 1
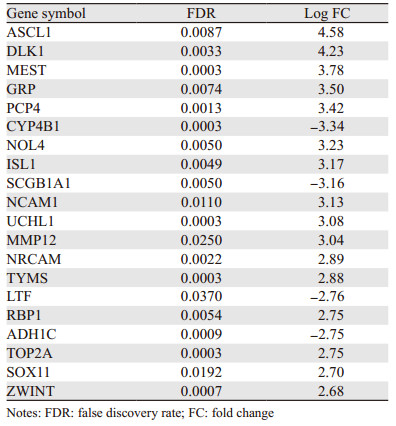
|
依据基因编码的蛋白质在细胞中的作用,GO分析将DEGs功能注释的结果分为三类:生物过程、细胞组分和分子功能。通过Cytoscape3.6.0软件绘制GO富集可视互作网络,其中细胞组分和分子功能部分结果见图 1。通过DAVID进行在线分析,筛选FDR < 0.05的结果,见图 2。在生物过程中,包括细胞分裂、G1/S有丝分裂细胞周期的转变、DNA复制、有丝分裂核分裂、有丝分裂姐妹染色单体分离、DNA复制启动、姐妹染色单体粘连、p53信号转导调控、有丝分裂胞质分裂、有丝分裂纺锤体组织、DNA修复、重组端粒维持、炎性反应的调节;在细胞组分中,包括核质、细胞质、细胞外泌体、细胞外空间、主轴、核、着丝粒、染色体、中体、浓缩染色体着丝粒以及微管;在分子功能中,包括与蛋白结合、与微管结合、与钙依赖性蛋白结合、与受损DNA结合、与染色质结合、与ATP结合以及与酶结合。

|
| A: the network diagram of the cellular components; B: the network diagram of molecular function 图 1 GO分析可视化网络 Figure 1 Visualization network of GO analysis |

|
| 图 2 DEGs的GO功能富集分析结果 Figure 2 GO function enrichment analysis results of differentially-expressed genes |
通过对肿瘤组织和正常肺组织的DEGs通路富集分析,结果表明DEGs主要集中在12条信号转导通路上,包括细胞周期、卵母细胞减数分裂、DNA复制、错配修复、碱基切除修复、核苷酸切除修复、补体和凝血级联反应、小细胞肺癌、癌症通路、P53信号通路、HTLV-I感染、病毒致癌作用等,见表 2。
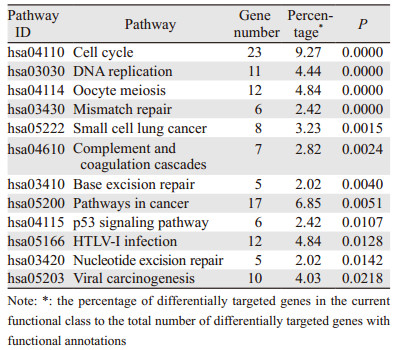
|
通过Cytoscape3.6.0软件构建248个DEGs的蛋白互作网络,剔除孤立节点后,筛选出的DEGs共同构成结构复杂的多中心互作网络。通过MCODE插件筛选出核心基因,明确核心基因有58个,其中节点度最高的6个基因如下:TOP2A(degree=95)、PCNA(degree=94)、RFC4(degree=83)、FEN1(degree=75)、CCNA2(degree=74)和MCM2(degree=74),见图 3。

|
| Red: the gene expressions were up-regulated; Light blue: the gene expressions were down-regulated. The darker the color was, the smaller the FDR value was; the larger the circle was, the larger the degree value was. 图 3 DEGs的蛋白网络互作图 Figure 3 Protein network interaction map of DEGs |
利用Kaplan-Meier分析6个核心基因与肺癌预后之间的关系,结果显示TOP2A、RFC4、FEN1、CCNA2、MCM2等基因高表达组的中位生存时间明显短于低表达组,差异有统计学意义(P < 0.5);PCNA高表达组与低表达组之间中位生存时间比较差异无统计学意义(P > 0.5),见图 4。

|
| 图 4 核心基因对肺癌患者中位生存时间的影响 Figure 4 Effect of core gene on prognosis of lung cancer patients analyzed with Kaplan-Meier online tool |
SCLC具有恶性程度高、早期广泛转移等特点,广泛期患者的生存时间仅为8至13月,2年生存率约为5%[6]。SCLC的耐药性及高复发率主要是由于基因高突变率和基因组不稳定性造成[7],研究表明P53和RB1是SCLC中突变频率最高的基因,突变频率分别为85%和57%,P53和RB1的突变预示着预后不良[8]。SCLC发病的分子机制尚未明确,迫切需要找到可用的潜在生物标志物,而生物信息学能够帮助我们探索SCLC基因层面发生的变化、识别潜在的生物标志物。
本研究通过GEO数据库检索获得GSE6044和GSE40275两个基因芯片数据集,原数据集均纳入肺癌的各个病理类型进行研究,但未对SCLC进行单独讨论且样本量较少。我们将两个数据集进行整合分析,扩大样本量,深入探讨SCLC发生、发展的机制以及潜在的治疗靶点。利用生物信息学进行SCLC差异基因筛选,共获得248个DEGs,包括172个高表达基因和76个低表达基因。在GO和KEGG富集分析中,高表达基因主要富集在细胞周期、DNA复制、卵母细胞减数分裂、癌症途径等通路,而低表达基因主要富集在错配修复、碱基切除修复等通路。细胞损伤的累积常常导致细胞的异常增殖和基因组不稳定[9]。细胞周期的失控是恶性肿瘤的标志,在肿瘤的致癌或进展过程中发挥重要作用[10]。DNA损伤主要通过错配修复、碱基切除修复、核苷酸切除修复等途径进行修复,相关基因的异常表达和甲基化导致DNA损伤的积累和肿瘤的发生[11]。此外,本研究发现部分DEGs还富集在补体和凝血级联反应、p53信号通路、化学致癌作用等通路。最近研究表明,补体系统是原发肿瘤和转移靶向器官出现免疫抑制状态的重要调控途径[12-13]。虽然目前相关文献还未明确阐述补体在恶性肿瘤中的作用,但有研究指出补体在肿瘤微环境中能够招募及诱导免疫抑制细胞的聚集[14];此外,在肺癌小鼠模型中抑制补体的表达和阻断程序性细胞死亡因子1具有协同抗肿瘤作用,可延缓肿瘤进展[15]。
通过Cytoscape3.6.0软件构建DEGs的蛋白互作网络图,发现6个高表达的核心基因,Kaplan-Meier分析显示TOP2A、RFC4、FEN1、CCNA2和MCM2的高表达预示着患者的预后不良。TOP2A是一种控制DNA拓扑状态的酶,可催化双链DNA断裂并促进有丝分裂期间的基因转录[16]。TOP2A通过zeste同源物2的增强子参与表观遗传的调控,TOP2A的异常表达与肿瘤的恶性特征相关[17]。在KEGG通路富集分析中MCM2、PCNA和CCNA2富集于细胞周期相关通路。MCM2亦称为微小染色体维持蛋白2,作为恶性肿瘤增殖的生物标志物,是ATP酶活性位点之一,其促进ATP构象变化并驱动DNA复制起点处的DNA解旋[18]。PCNA定位于细胞核,是细胞增殖过程中关键成分之一,在S1期高表达,是DNA复制过程中DNA聚合酶的重要辅助因子[19]。EGF、HGFL等细胞生长因子通过C-Abl激酶提高PCNA的表达水平从而促进细胞增殖[20]。PCNA在肺癌、乳腺癌等肿瘤中的表达水平明显升高,能够促进肿瘤转移并与患者的生存率相关[21]。CCNA2通过与蛋白激酶CDK2和CDK1的结合参与有丝分裂G1/S和G2/M期,促进细胞增殖[22]。另外,本研究发现RFC4和FEN1富集于DNA复制相关通路。RFC4是复制因子C家族成员之一,主要参与DNA复制和细胞周期检查点的调控,其高表达与肿瘤分化程度、TNM分期和预后相关[23]。FEN1是一种结构特异性多功能核酸酶,在真核细胞的DNA复制和DNA代谢途径中具有重要作用,被认为是维持基因组稳定性和防止肿瘤发生的关键基因[24];据报道FEN1在肿瘤细胞中突变导致核酸酶活性降低,并且70%的小鼠敲入突变的FEN1后在多个器官中发生恶性肿瘤[25];同样,FEN1在肺癌组织中高度过表达[26]。
综上所述,本研究通过GEO数据库中的芯片数据深入挖掘DEGs及其靶蛋白,结果表明相关基因在SCLC的发生、转移中起着重要作用,在基因层面为寻找新的分子靶点提供了一定的支持,也为实现SCLC的精准治疗提供了一个新思路,但还需进一步实验验证相关结果。
作者贡献
毛昀、李林潞:论文设计、书写和数据分析
薛鹏、徐芃芃:文献查阅和图表制作
褚雪镭、姜朋媛、蔡亚芳:参与论文书写、校对
朱世杰:论文设计、修改
| [1] | Bray F, Ferlay J, Soerjomataram I, et al. Global cancer statistics 2018: Globocan estimates of incidence and mortality worldwide for 36 cancers in 185 countries[J]. CA Cancer J Clin, 2018, 68(6): 394–424. DOI:10.3322/caac.21492 |
| [2] | Xie D, Marks R, Zhang M, et al. Nomograms predict overall survival for patients with small-cell lung cancer incorporating pretreatment peripheral blood markers[J]. J Thorac Oncol, 2015, 10(8): 1213–1220. DOI:10.1097/JTO.0000000000000585 |
| [3] | Waqar SN, Morgensztern D. Treatment advances in small cell lung cancer (sclc)[J]. Pharmacol Ther, 2017, 180: 16–23. DOI:10.1016/j.pharmthera.2017.06.002 |
| [4] | Sakre N, Wildey G, Behtaj M, et al. RICTOR amplification identifies a subgroup in small cell lung cancer and predicts response to drugs targeting mTOR[J]. Oncotarget, 2016, 8(4): 5992–6002. |
| [5] | Rohrbeck A, Neukirchen J, Rosskopf M, et al. Gene expression profiling for molecular distinction and characterization of laser captured primary lung cancers[J]. J Transl Med, 2008, 6: 69. DOI:10.1186/1479-5876-6-69 |
| [6] | Travis WD. Update on small cell carcinoma and its differentiation from squamous cell carcinoma and other non-small cell carcinomas[J]. Mod Pathol, 2012, 25(Suppl 1): S18–S30. |
| [7] | Karachaliou N, Pilotto S, Lazzari C, et al. Cellular and molecular biology of small cell lung cancer: An overview[J]. Transl Lung Cancer Res, 2016, 5(1): 2–15. |
| [8] | Sundaresan V, Lin VT, Liang F, et al. Significantly mutated genes and regulatory pathways in sclc-a meta-analysis[J]. Cancer Genet, 2017, 216-217: 20–28. DOI:10.1016/j.cancergen.2017.05.003 |
| [9] | Dominguez-Brauer C, Thu K, Mason J, et al. Targeting mitosis in cancer: Emerging strategies[J]. Mol Cell, 2015, 60(4): 524–536. DOI:10.1016/j.molcel.2015.11.006 |
| [10] | Penna LS, Henriques JAP, Bonatto D. Anti-mitotic agents: Are they emerging molecules for cancer treatment?[J]. Pharmacol Ther, 2017, 173: 67–82. DOI:10.1016/j.pharmthera.2017.02.007 |
| [11] | Hansen LT, Thykjaer T, Orntoft TF, et al. The role of mismatch repair in small-cell lung cancer cells[J]. Eur J Cancer, 2003, 39(10): 1456–1467. DOI:10.1016/S0959-8049(03)00306-X |
| [12] | Reis ES, Mastellos DC, Ricklin D, et al. Complement in cancer: Untangling an intricate relationship[J]. Nat Rev Immunol, 2018, 18(1): 5–18. |
| [13] | Sharma SK, Chintala NK, Surya Kumari V, et al. Pulmonary alveolar macrophages contribute to the premetastatic niche by suppressing antitumor t cell responses in the lungs[J]. J Immunol, 2015, 194(11): 5529–5538. DOI:10.4049/jimmunol.1403215 |
| [14] | Afshar-Kharghan V. The role of the complement system in cancer[J]. J Clin Invest, 2017, 127(3): 780–789. DOI:10.1172/JCI90962 |
| [15] | Ajona D, Ortiz-Espinosa S, Moreno H, et al. A combined pd-1/c5a blockade synergistically protects against lung cancer growth and metastasis[J]. Cancer Discov, 2017, 7(7): 694–703. DOI:10.1158/2159-8290.CD-16-1184 |
| [16] | Wu KZ, Wang GN, Fitzgerald J, et al. DDK dependent regulation of TOP2A at centromeres revealed by a chemical genetics approach[J]. Nucleic Acids Res, 2016, 44(18): 8786–8798. DOI:10.1093/nar/gkw626 |
| [17] | Kirk JS, Schaarschuch K, Dalimov Z, et al. Top2a identifies and provides epigenetic rationale for novel combination therapeutic strategies for aggressive prostate cancer[J]. Oncotarget, 2015, 6(5): 3136–3146. |
| [18] | Cheung CHY, Hsu CL, Chen KP, et al. Mcm2-regulated functional networks in lung cancer by multi-dimensional proteomic approach[J]. Sci Rep, 2017, 7(1): 13302. DOI:10.1038/s41598-017-13440-x |
| [19] | Siddiqui K, On KF, Diffley JF. Regulating DNA replication in eukarya[J]. Cold Spring Harb Perspect Biol, 2013, 5(9): pii: a012930. DOI:10.1101/cshperspect.a012930 |
| [20] | Zhao H, Chen MS, Lo YH, et al. The ron receptor tyrosine kinase activates c-abl to promote cell proliferation through tyrosine phosphorylation of pcna in breast cancer[J]. Oncogene, 2014, 33(11): 1429–1437. DOI:10.1038/onc.2013.84 |
| [21] | Hoffmann S, Smedegaard S, Nakamura K, et al. TRAIP is a PCNA-binding ubiquitin ligase that protects genome stability after replication stress[J]. J Cell Biol, 2016, 212(1): 63–75. DOI:10.1083/jcb.201506071 |
| [22] | Evan GI, Vousden KH. Proliferation, cell cycle and apoptosis in cancer[J]. Nature, 2001, 411(6835): 342–348. DOI:10.1038/35077213 |
| [23] | Xiang J, Fang L, Luo Y, et al. Levels of human replication factor c4, a clamp loader, correlate with tumor progression and predict the prognosis for colorectal cancer[J]. J Transl Med, 2014, 12: 320. DOI:10.1186/s12967-014-0320-0 |
| [24] | Balakrishnan L, Bambara RA. Flap endonuclease 1[J]. Annu Rev Biochem, 2013, 82: 119–138. DOI:10.1146/annurev-biochem-072511-122603 |
| [25] | Zheng L, Dai H, Zhou M, et al. Fen1 mutations result in autoimmunity, chronic inflammation and cancers[J]. Nat Med, 2007, 13(7): 812–819. DOI:10.1038/nm1599 |
| [26] | Nikolova T, Christmann M, Kaina B. Fen1 is overexpressed in testis, lung and brain tumors[J]. Anticancer Res, 2009, 29(7): 2453–2459. |