 2019, Vol. 46
2019, Vol. 46文章信息
- 肿瘤中SWI/SNF复合物亚基变异的作用及其相关治疗策略进展
- Role of SWI/SNF Complex Subunit Abnormality in Tumors and Progress of Related Therapeutic Strategies
- 肿瘤防治研究, 2019, 46(7): 644-649
- Cancer Research on Prevention and Treatment, 2019, 46(7): 644-649
- http://www.zlfzyj.com/CN/10.3971/j.issn.1000-8578.2019.18.1871
- 收稿日期: 2018-12-07
- 修回日期: 2019-02-02
引用本文 |



2. 210002 南京,解放军南京总医院病理科
2. Department of Pathology, Nanjing General Hospital of PLA, Nanjing 210002, China
染色质重构是基因表达动态调控的重要机制之一,主要由不同的蛋白质/蛋白质复合物完成[1]。SWI/SNF复合物是首次在酵母中发现的影响交配型转换的基因复合物之一,转录调控因子等蛋白将SWI/SNF复合物募集到DNA区域,沿DNA移动并通过变构核小体修改DNA的可访问性或可及性(重塑染色质),通过转录调节因子与暴露DNA的结合实现基因表达调控(抑制或者激活)。其亚基基因在20%以上的恶性肿瘤中发生突变[2]。
1 SWI/SNF复合物及其功能哺乳动物SWI/SNF复合物由多个亚基组成,包括两个催化亚基BRM/SMARCA2和BRG1/SMARCA4,以及一组称为BRG1/BRM相关因子的蛋白质(是SWI/SNF复合物与DNA或蛋白质结合所必需的亚基)[2]。SWI/SNF复合物主要包含两类:BRG1/BRM相关因子复合物(BRG1/BRM associated factor, BAF)及多溴相关BAF复合物(polybromo-associated BAF, PBAF)。BAF及PBAF均包括三个核心亚基(SMARCB1、SMARCC1及SMARCC 2),辅助调节亚基存在差异,BAF主要包括ARID1A/1B、DPF1/2/3、SS18、SMARCE1、SMARCD1/D2/D3、ACTL6A及BRD9,PBAF主要包括ARID2、PBRM1、PHF10、SMARCE1、SMARCD1/D2/D3、ACTL6A及BRD7。SMARCA2及SMARCA4含有6个保守结构域:QLQ结构域、富含脯氨酸的结构域、小解旋酶/SANT相关结构域、DNA依赖性ATP酶结构域、视网膜母细胞瘤(RB)结合结构域(LxCxE)和Bromo结构域。Bromo结构域与乙酰化组蛋白相互作用,参与SWI/SNF复合物与DNA的结合和稳定性。LxCxE结构域与RB肿瘤抑制基因家族的成员结合,而QLQ结构域与蛋白质-蛋白质相互作用有关。最后,解旋酶和DExDc结构域分离DNA双链,需ATP水解[3]。但这两种ATP水解酶在功能上存在显著差别,并不能彼此弥补。INI 1/SMARCB 1、BAF 155/SMARCC1和BAF170/SMARCC2称为“核心亚基”,其对于SMARCA2或SMARCA4的ATP依赖染色质重塑活性是必需的,主要参与双链断裂和核苷酸切除修复。SWI/SNF复合物还有7~10个辅助调节亚基,靶向特定的DNA或基因位点,负责不同复合物靶向的特异性基因组。BAF复合物包含ARID1A/B,PBAF复合物包含PBRM1、ARID2、BRD7和PHF10[4]。其含有与DNA或组蛋白相互作用所需的特定结构域(溴结构域、染色质结构域、DNA结合结构域、ARID及Zing finger等)[2-4]。
SWI/SNF亚基不仅与启动子结合,而且与其他调控区域(如增强子和DNA复制起始区)紧密结合。此外,SWI/SNF可结合/共沉淀许多蛋白质,在细胞周期、细胞骨架和染色体组织等过程中发挥作用[3]。表明SWI/SNF复合物的功能比单纯的转录调控更为广泛。
2 SWI/SNF复合物亚基变异在肿瘤中的作用现针对各个SWI/SNF复合物亚基变异进行分析,见表 1[3, 5]。
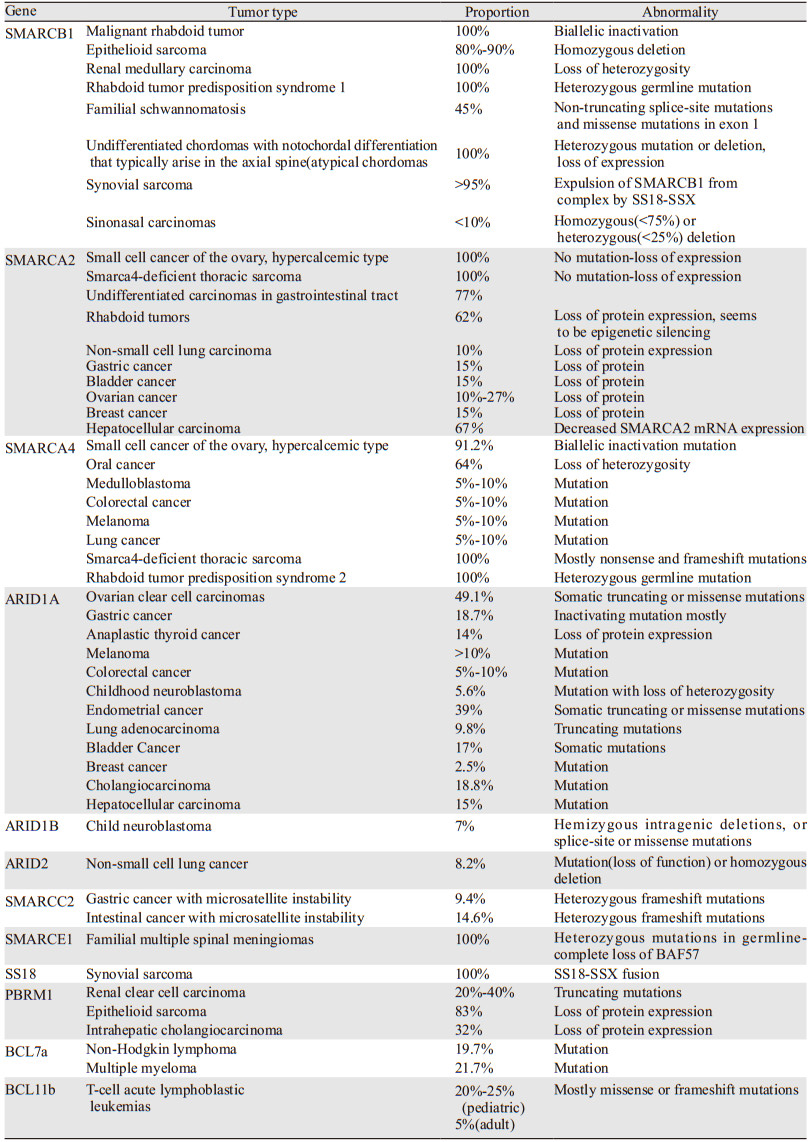
SMARCA2和SMARCA4虽同为SWI/SNF的ATP水解酶,但两者在恶性肿瘤中的突变谱明显不同。SMARCA4和SMARCA2在结直肠癌、浆液性卵巢癌、胰腺癌、肝细胞癌、肺癌和乳腺癌、胶质瘤、黑色素瘤和鳞状细胞癌中均有不同频率的突变,在卵巢透明细胞癌、肾癌、血液系统肿瘤和髓母细胞瘤中存在SMARCA4突变但无SMARCA2突变[5-6],大多数突变位于解旋酶催化结构域。SMARCA4是继ARID1A之后恶性肿瘤中第二频繁突变的SWI/SNF基因,其作用比SMARCA2更重要。
在卵巢高钙血症型小细胞癌(small cell carcinoma of the ovary, SCCOHT)中均有SMARCA2蛋白缺失[7],常发生表观基因改变。SMARCA2缺失可发生在横纹肌样瘤、乳腺癌、肺癌、食道癌、膀胱癌、胃癌和卵巢癌中[7-8]。SMARCA2突变非常罕见,但可见于腺样囊性癌(5%)[9]。SMARCA4和SMARCA2是SWI/SNF的相对互斥亚基,在SMARCA4缺陷肿瘤中,SMARCA2上调,可补充SMARCA4的功能丢失[4]。但在SCCOHT和SMARCA4缺陷性胸部肉瘤(SMARCA4-deficient thoracic sarcomas, SMARCA4-DTS)中SMARCA4和SMARCA2表达同时丢失[10],与在其他SMARCA4缺陷肿瘤中的合成致死关系不同。
在SCCOHT中均有SMARCA4基因突变。SCCOHT是双等位SMARCA4失活突变(包括截短、移码或缺失),近一半的SCOOHT在种系细胞中检测出SMARCA4突变,并可能表现为横纹肌样瘤易感性综合征[11]。SMARCA4-DTS也与重复性SMARCA4突变相关,导致SMARCA4功能丧失[12]。
催化亚基突变也出现在胃肠道低分化癌(undifferentiated gastrointestinal carcinoma, UGC)中,包括:结肠、小肠、胃和远端食道,主要显示SMARCB1或SMARCA4失活,也有SMARCA2和ARID1A改变。SMARCA2和SMARCB1是UGC中最常见的SWI/SNF突变亚基(分别为77%和50%)。所有SMARCB1突变的病例也有SMARCA2突变,而SMARCA2和SMARCA4突变相互排斥,SMARCB1和SMARCA4之间也是如此[3]。
2.2 核心亚基SMARCB1几乎在所有恶性横纹肌样肿瘤(malignant rhabdoid tumor, MRT)中都存在基因缺失或截短突变,是MRT中唯一的重复性遗传异常[13]。在家族性神经瘤病和脑膜瘤中有SMARCB1双等位基因丢失。80%以上的上皮样肿瘤中也存在SMARCB1蛋白丢失(主要因纯合子缺失)。而其他两个核心亚基(SMARCC1和SMARCC2)却鲜有突变[5]。
在MRT中,SMARCB1缺失增加了多梳抑制复合物2(polycomb repressive complex 2, PRC2)亚基EZH2的丰度,EZH2高表达使PRC2不能被置换,导致DNA甲基化,从而抑制肿瘤抑制基因CDKN2A,导致增殖增加[14]。
在肾髓质癌[15]中存在不平衡易位后的SMARCB1截短。SMARCB1功能丧失可诱导细胞周期蛋白D1,导致细胞周期的G1期细胞增殖。SMARCB1缺陷约占鼻窦癌的10%[16]。与MRT一样,其基因组高度稳定,表明SMARCB1丢失可能是其发生的驱动因素。低分化小儿脊索瘤存在由染色体22q11缺失引起的SMARCB1缺失[17]。
2.3 辅助调节亚基在家族性脊膜瘤中,SMARCE1杂合突变并完全丢失,并与透明细胞组织学形态相关[18],更具侵袭性,较易扩散转移,可能与SMARCE1作用于类固醇激素反应有关[19]。在透明细胞肾癌(约40%)、上皮样肉瘤(约83%)和胆管癌(约32%)中存在PBRM1失活突变或缺失。14%的HCV相关肝细胞肝癌、小部分恶性黑色素细胞瘤和肺癌有ARID2失活突变[5]。
ARID1A是肿瘤中突变频率最高的SWI/SNF基因,一项约3 000份肿瘤标本的大型研究发现,其在10%以上的肿瘤中存在丢失:胃癌(14%)、间变性甲状腺癌(14%)、低级别和高级别子宫内膜样癌(分别占29%和39%)、子宫癌(浆液癌18%、癌肉瘤14%)[5]。并在肝细胞癌[20]、肺腺癌[21]、膀胱癌[22]或胆管癌[23]中存在突变。约50%的卵巢透明细胞癌及近40%的子宫内膜癌存在ARID1A突变,其也与良性子宫内膜异位症癌变相关[24]。ARID1A在肿瘤中常发生截短突变,导致C末端结合区的缺失,破坏复合物的组合。ARID1A丢失并不能通过ARID1B的表达增多加以补偿,且在大多数肿瘤中ARID1B表达比ARID1A更低[25]。
SMARCC2突变很罕见,但可见于微卫星不稳定的胃癌和结直肠癌[26]。SKARCC2外显子8的重复序列是移码突变的热点,分别存在于胃癌(9%)和结直肠癌(15%)中。该突变产生终止密码子,导致SMARCC2的功能丧失。
2.4 滑膜肉瘤SS18移位SS18首先在滑膜肉瘤(synovial sarcoma, SS)中被鉴定,t(X; 18)(p11.2; q11.2)易位引起18号染色体上的SS18与X染色体上的SSX1、SSX2或SSX4融合[27],导致SS18 C末端与SSX蛋白的78个C末端氨基酸融合。SS18-SSX融合蛋白与正常SS18亚基竞争结合到SWI/SNF复合物中。可能由于融合蛋白体积较大,SMARCB1亚基从滑膜肉瘤BAF(ssBAF)复合物中被置换。通常,SMARCB1被排出及降解导致功能丧失,然而,SW1/SNF复合物中的SS18-SSX进入整合导致复合物功能增强,即染色质占据能力增强和PRC被强力驱逐置换。融合蛋白的SSX与特定DNA位点结合可导致ssBAF定向到其他的基因组位点[28],激活SOX2致癌基因。该机制使SWI/SNF复合物具有了致癌活性。
3 其他SWI/SNF相关肿瘤发生机制SWI/SNF复合物可通过与长链非编码RNA(lncRNA)相互作用参与肿瘤发生。其机制是:lncRNA直接与SWI/SNF复合物相互作用,拮抗其活性,或使SWI/SNF复合物在某些特定位点募集[29],促使肿瘤发生。
最近研究成果包括:(1)发现SWI/SNF复合物在谱系特异性基因增强子调节中的作用[30]。SWI/SNF复合物直接与p300组蛋白乙酰转移酶结合,从而调节靶基因上H3K27Ac(组蛋白H3K27乙酰化)的活性。这种SWI/SNF调节在典型的远端增强子中显示出强活性,主要参与分化和发育的基因。提示SWI/SNF亚基缺失(如SMARCB1、ARID1A)可通过该途径诱导肿瘤发生; (2)如前所述,SWI/SNF复合物(如SMARCB1)与PRC2的EZH2之间存在特异性拮抗关系,多梳复合物是SWI/SNF的关键非核小体底物。EZH2负责组蛋白H3第27位赖氨酸的三甲基化,这是与基因组转录抑制相关的标记[14]。
4 SWI/SNF复合物亚基驱动相关肿瘤的新型治疗策略探讨在临床上,对异常缺失的基因产物进行恢复很难实现,因此,对于基因表达缺失驱动肿瘤的治疗仍然存在很大的挑战。但SWI/SNF亚基异常与其他致癌基因异常共同发生,因此,利用特异性的合成致死关系是其治疗的有效途径。合成致死关系是导致细胞适应性丧失的两种遗传事件(如突变)的组合,即两者同时发生将会导致细胞死亡。目前,对于SWI/SNF复合物亚基之间或SWI/SNF亚基缺失与异常激活的致癌信号途径之间的合成致死关系已经成为精准治疗的热门领域。目前主要如下。
4.1 SWI/SNF复合物旁系同源亚基内部之间SWI/SNF亚基缺失肿瘤会表现出对其他旁系同源亚基独特的强依赖性,以维持其增殖适应性。如SMARCA4-SMARCA2及ARID1A-ARID1B。在SMARCA4缺陷非小细胞肺癌(NSCLC)细胞系中,RNAi文库合成致死筛选发现,SMARCA2为癌细胞系增殖所需的最强合成致死依赖性亚基[31]。可使用靶生物分子的配体依赖性蛋白质降解技术特异性降解亚基,直接靶向SWI/SNF ATP酶和溴结构域[32]。
4.2 SMARCB1表达缺失和EZH2抑制SMARCB1表达缺失和PRC2抑制之间存在合成致死关系,EZH2是PRC2的催化亚基。小分子介导EZH2抑制的临床前研究已在SMARCB1缺陷肿瘤中得到验证[33]。而且,特异性EZH2抑制小分子还可与其他关键效应信号抑制剂组合使用,以提高效果,如溴结构域或SHH信号抑制剂[32]。
4.3 SWI/SNF亚基缺失与其他下游依赖性信号通路在SWI/SNF亚基缺失肿瘤中,发现了多个其他下游依赖性信号通路的合成致死关系靶标,如YES1[34]、PARP [35]、ATR[36]、PIK3CA[37]、KRAS[38]、BRAF[3]、Topo Ⅱ[39]及细胞周期蛋白D1[40]等。
SWI/SNF复合物与DNA损伤修复有关。SWI/SNF亚基缺失癌细胞对DNA修复途径抑制剂的易感性增强。PIK3/AKT途径在该领域具有重要意义。基于shRNA的筛选研究发现,PIK3CA(p110)和PIK3R2(p85)的两个催化亚基在ARID1A缺陷癌细胞中显示很高的脆弱性。抑制PIK3途径药物可能是易实现的临床选择。事实上,一些针对PIK3/AKT途径节点的制剂已快速转向FDA批准[41]。SMARCA4表达缺失与对拓扑异构酶Ⅱ抑制剂的耐受性相关,EZH2抑制剂与依托泊苷(一种拓扑异构酶Ⅱ(Topo Ⅱ)抑制剂)在SMARCA4缺失肿瘤中具有协同抗肿瘤作用[42]。SMARCB1失活可导致细胞周期蛋白D1上调。目前一种细胞周期蛋白依赖性激酶(CDK)4/6抑制剂ribociclib(LEE011)正在进行阶段1/2临床试验[40]。
4.4 其他目前,针对滑膜肉瘤中致癌性SS18-SSX特异性降解或针对SS18-SSX的合成致死关系的治疗途径还在研究中。此外,SWI/SNF亚基还可作为治疗反应的预测因子。如类固醇治疗对小儿急性淋巴细胞白血病的反应与3个SWI/SNF亚基(SMARCB1、SMARCA4、ARID1A)的表达水平相关:低表达与较高的治疗反应相关[43]。SMARCE1表达可用作卵巢癌和肺癌药物反应的标志物:卵巢癌对顺铂、多柔比星和5-氟尿嘧啶的敏感度可能与低SMARCE1表达有关[44],低SMARCE1表达也与非小细胞肺癌的MET和ALK抑制剂耐药相关[45]。
5 总结SWI/SNF复合物在细胞信号调控等多个方面发挥了重要作用,并且,其亚基异常涉及较多恶性肿瘤的发生发展,因此,对其复杂的作用机制及临床应用开发研究很有意义,如进一步研究SWI/SNF复合物异常对恶性肿瘤预后的影响、其表达缺失的具体机制(如SNP、插入缺失、拷贝数变异、体细胞DNA突变、甲基化/染色质结构/基因表达水平)及致癌机制(包括单个及多个亚基),基因组学时代的来临也使系统研究SWI/SNF复合物异常涉及的信号通路、合成致死筛选及具体机制成为可能,从而为此类肿瘤的靶向精准治疗带来希望。
作者贡献
郭庆:文献整理及撰写
饶秋、周志毅:指导论文撰写
| [1] | Muchardt C, Yaniv M. ATP-dependent chromatin remodelling: SWI/SNF and Co. are on the job[J]. J Mol Biol, 1999, 293(2): 187–198. DOI:10.1006/jmbi.1999.2999 |
| [2] | Mashtalir N, D'Avino AR, Michel BC, et al. Modular organization and assembly of SWI/SNF family chromatin remodeling complexes[J]. Cell, 2018, 175(5): 1272–1288. DOI:10.1016/j.cell.2018.09.032 |
| [3] | Arnaud O, Le Loarer F, Tirode F. BAFfling pathologies: Alterations of BAF complexes in cancer[J]. Cancer Lett, 2018, 419: 266–279. DOI:10.1016/j.canlet.2018.01.046 |
| [4] | Kadoch C, Crabtree GR. Mammalian SWI/SNF chromatin remodeling complexes and cancer: Mechanistic insights gained from human genomics[J]. Sci Adv, 2015, 1(5): e1500447. DOI:10.1126/sciadv.1500447 |
| [5] | Savas S, Skardasi G. The SWI/SNF complex subunit genes: Their functions, variations, and links to risk and survival outcomes in human cancers[J]. Crit Rev Oncol Hematol, 2018, 123: 114–131. DOI:10.1016/j.critrevonc.2018.01.009 |
| [6] | Raab JR, Runge JS, Spear CC, et al. Co-regulation of transcription by BRG1 and BRM, two mutually exclusive SWI/SNF ATPase subunits[J]. Epigenetics Chromatin, 2017, 10(1): 62. DOI:10.1186/s13072-017-0167-8 |
| [7] | Karnezis AN, Wang Y, Ramos P, et al. Dual loss of the SWI/SNF complex ATPases SMARCA4/BRG1 and SMARCA2/BRM is highly sensitive and specific for small cell carcinoma of the ovary, hypercalcaemic type[J]. J Pathol, 2016, 238(3): 389–400. DOI:10.1002/path.4633 |
| [8] | Ouyang X, Ye XL, Wei HB. BRM promoter insertion polymorphisms increase the risk of cancer: A meta-analysis[J]. Gene, 2017, 626: 420–425. DOI:10.1016/j.gene.2017.05.047 |
| [9] | Jagielska B, Sarnowska E, Rusetska N, et al. Advanced adenoid cystic carcinoma (ACC) is featured by SWI/SNF chromatin remodeling complex aberrations[J]. J Cancer Res Clin Oncol, 2018, 145(1): 201–211. |
| [10] | Kuwamoto S, Matsushita M, Takeda K, et al. SMARCA4-deficient thoracic sarcoma: report of a case and insights into how to reach the diagnosis using limited samples and resources[J]. Hum Pathol, 2017, 70: 92–97. DOI:10.1016/j.humpath.2017.05.024 |
| [11] | Lang JD, Hendricks WPD. Identification of driver mutations in rare cancers: the role of SMARCA4 in small cell carcinoma of the ovary, hypercalcemic type (SCCOHT)[J]. Methods Mol Biol, 2018, 1706: 367–379. DOI:10.1007/978-1-4939-7471-9 |
| [12] | Yoshida A, Kobayashi E, Kubo T, et al. Clinicopathological and molecular characterization of SMARCA4-deficient thoracic sarcomas with comparison to potentially related entities[J]. Mod Pathol, 2017, 30(6): 797–809. |
| [13] | Pawel BR. SMARCB1-deficient tumors of childhood: a practical guide[J]. Pediatr Dev Pathol, 2018, 21(1): 6–28. |
| [14] | Kadoch C, Williams RT, Calarco JP, et al. Dynamics of BAF-Polycomb complex opposition on heterochromatin in normal and oncogenicstates[J]. Nat Genet, 2017, 49(2): 213–222. DOI:10.1038/ng.3734 |
| [15] | Calderaro J, Masliah-Planchon J, Richer W, et al. Balanced translocations disrupting SMARCB1 are hallmark recurrent genetic alterations in renal medullary carcinomas[J]. Eur Urol, 2016, 69(6): 1055–1061. DOI:10.1016/j.eururo.2015.09.027 |
| [16] | Laco J, Chmelařová M, Vošmiková H, et al. SMARCB1/INI1-deficient sinonasal carcinoma shows methylation of RASSF1 gene: A clinicopathological, immunohistochemical and molecular genetic study of a recently described entity[J]. Pathol Res Pract, 2017, 213(2): 133–142. DOI:10.1016/j.prp.2016.10.012 |
| [17] | Hasselblatt M, Thomas C, Hovestadt V, et al. Poorly differentiated chordoma with SMARCB1/INI1 loss: a distinct molecular entity with dismalprognosis[J]. Acta Neuropathol, 2016, 132(1): 149–151. DOI:10.1007/s00401-016-1574-9 |
| [18] | Pathmanaban ON, Sadler KV, Kamaly-Asl ID, et al. Association of genetic predisposition with solitary schwannoma or meningioma in children and young adults[J]. JAMA Neurol, 2017, 74(9): 1123–1129. DOI:10.1001/jamaneurol.2017.1406 |
| [19] | Lomelí H, Castillo-Robles J. The developmental and pathogenic roles of BAF57, a special subunit of the BAF chromatin-remodeling complex[J]. FEBS Let, 2016, 590(11): 1555–1569. DOI:10.1002/1873-3468.12201 |
| [20] | Ikeda S, Tsigelny IF, Skjevik ÅA, et al. Next-generation sequencing of circulating tumor DNA reveals frequent alterations in advanced hepatocellular carcinoma[J]. Oncologist, 2018, 23(5): 586–593. DOI:10.1634/theoncologist.2017-0479 |
| [21] | Lissanu Deribe Y, Sun Y, Terranova C, et al. Mutations in the SWI/SNF complex induce a targetable dependence on oxidative phosphorylation in lung cancer[J]. Nat Med, 2018, 24(7): 1047–1057. DOI:10.1038/s41591-018-0019-5 |
| [22] | Agarwal N, Pal SK, Hahn AW, et al. Characterization of metastatic urothelial carcinoma via comprehensive genomic profiling of circulating tumor DNA[J]. Cancer, 2018, 124(10): 2115–2124. DOI:10.1002/cncr.v124.10 |
| [23] | Simbolo M, Vicentini C, Ruzzenente A, et al. Genetic alterations analysis in prognostic stratified groups identified TP53 and ARID1A as poor clinical performance markers in intrahepatic cholangiocarcinoma[J]. Sci Rep, 2018, 8(1): 7119. DOI:10.1038/s41598-018-25669-1 |
| [24] | Lakshminarasimhan R, Andreu-Vieyra C, Lawrenson K, et al. Down-regulation of ARID1A is sufficient to initiate neoplastic transformation along with epigenetic reprogramming in non-tumorigenic endometriotic cells[J]. Cancer Lett, 2017, 401: 11–19. DOI:10.1016/j.canlet.2017.04.040 |
| [25] | Mashtalir N, D'Avino AR, Michel BC, et al. Modular organization and assembly of SWI/SNF family chromatin remodeling complexes[J]. Cell, 2018, 175(5): 1272–1288. DOI:10.1016/j.cell.2018.09.032 |
| [26] | Kim YS, Jeong H, Choi JW, et al. Unique characteristics of ARID1A mutation and protein level in gastric and colorectal cancer: A meta-analysis[J]. Saudi J Gastroenterol, 2017, 23(5): 268–274. DOI:10.4103/sjg.SJG_184_17 |
| [27] | Morgan MA, Shilatifard A. Epigenetic ConFUSION: SS18-SSX fusion rewires BAF complex to activate bivalent genes in synovial sarcoma[J]. Cancer Cell, 2018, 33(6): 951–953. DOI:10.1016/j.ccell.2018.05.011 |
| [28] | McBride MJ, Pulice JL, Beird HC, et al. The SS18-SSX fusion oncoprotein hijacks BAF complex targeting and function to drive synovial sarcoma[J]. Cancer Cell, 2018, 33(6): 1128–1141. DOI:10.1016/j.ccell.2018.05.002 |
| [29] | Tang Y, Wang J, Lian Y, et al. Linking long non-coding RNAs and SWI/SNF complexes to chromatin remodeling in cancer[J]. Mol Cancer, 2017, 16(1): 42. DOI:10.1186/s12943-017-0612-0 |
| [30] | Alver BH, Kim KH, Lu P, et al. The SWI/SNF chromatin remodeling complex is required for maintenance of lineage specificenhancers[J]. Nat Commun, 2017, 8: 14648. DOI:10.1038/ncomms14648 |
| [31] | Hoffman GR, Rahal R, Buxton F, et al. Functional epigenetics approach identifies BRM/SMARCA2 as a critical synthetic lethal target in BRG1-deficient cancers[J]. Proc Natl Acad Sci U S A, 2014, 111(8): 3128–3133. DOI:10.1073/pnas.1316793111 |
| [32] | St Pierre R, Kadoch C. Mammalian SWI/SNF complexes in cancer: emerging therapeutic opportunities[J]. Curr Opin Genet Dev, 2017, 42: 56–67. DOI:10.1016/j.gde.2017.02.004 |
| [33] | Yamagishi M, Uchimaru K. Targeting EZH2 in cancer therapy[J]. Curr Opin Oncol, 2017, 29(5): 375–381. DOI:10.1097/CCO.0000000000000390 |
| [34] | Miller RE, Brough R, Bajrami I, et al. Synthetic lethal targeting of ARID1A-mutant ovarian clear cell tumors with dasatinib[J]. Mol Cancer Ther, 2016, 15(7): 1472–1484. DOI:10.1158/1535-7163.MCT-15-0554 |
| [35] | Shen J, Peng Y, Wei L, et al. ARID1A deficiency impairs the DNA damage checkpoint and sensitizes cells to PARP inhibitors[J]. Cancer Discov, 2015, 5(7): 752–767. DOI:10.1158/2159-8290.CD-14-0849 |
| [36] | Williamson CT, Miller R, Pemberton HN, et al. ATR inhibitors as a synthetic lethal therapy for tumours deficient in ARID1A[J]. Nat Commun, 2016, 7: 13837. DOI:10.1038/ncomms13837 |
| [37] | Samartzis EP, Gutsche K, Dedes KJ, et al. Loss of ARID1A expression sensitizes cancer cells to PI3K- and AKT-inhibition[J]. Oncotarget, 2014, 5(14): 5295–5303. |
| [38] | Wang SC, Nassour I, Xiao S, et al. SWI/SNF component ARID1A restrains pancreatic neoplasia formation[J]. Gut, 2018, pii: gutjnl-2017-315490. |
| [39] | Trotter KW, King HA, Archer TK. Glucocorticoid Receptor Transcriptional Activation via the BRG1-Dependent Recruitment of TOP2β and Ku70/86[J]. Mol Cell Biol, 2015, 35(16): 2799–2817. DOI:10.1128/MCB.00230-15 |
| [40] | Geoerger B, Bourdeaut F, DuBois SG, et al. A Phase I Study of the CDK4/6 Inhibitor Ribociclib (LEE011) in Pediatric Patients with Malignant Rhabdoid Tumors, Neuroblastoma, and Other Solid Tumors[J]. Clin Cancer Res, 2017, 23(10): 2433–2441. DOI:10.1158/1078-0432.CCR-16-2898 |
| [41] | Suvarna V, Murahari M, Khan T, et al. Phytochemicals and PI3K Inhibitors in Cancer-An Insight[J]. Front Pharmacol, 2017, 8: 916. DOI:10.3389/fphar.2017.00916 |
| [42] | Fillmore CM, Xu C, Desai PT, et al. EZH2 inhibition sensitizes BRG1 and EGFR mutant lung tumours to TopoII inhibitors[J]. Nature, 2015, 520(7546): 239–242. DOI:10.1038/nature14122 |
| [43] | Wu JN, Pinello L, Yissachar E, et al. Functionally distinct patterns of nucleosome remodeling at enhancers in glucocorticoid-treated acute lymphoblastic leukemia[J]. Epigenetics Chromatin, 2015, 8: 53. DOI:10.1186/s13072-015-0046-0 |
| [44] | Yamaguchi T, Kurita T, Nishio K, et al. Expression of BAF57 in ovarian cancer cells and drug sensitivity[J]. Cancer Sci, 2015, 106(4): 359–366. |
| [45] | Papadakis AI, Sun C, Knijnenburg TA, et al. SMARCE1 suppresses EGFR expression and controls responses to MET and ALK inhibitors in lung cancer[J]. Cell Res, 2015, 25(4): 445–458. |