 2018, Vol. 45
2018, Vol. 45文章信息
- 低糖增强肿瘤对二甲双胍的敏感度
- Low Glucose Enhances Sensitivity of Cancer to Metformin
- 肿瘤防治研究, 2018, 45(3): 125-130
- Cancer Research on Prevention and Treatment, 2018, 45(3): 125-130
- http://www.zlfzyj.com/CN/10.3971/j.issn.1000-8578.2018.18.0189
- 收稿日期: 2018-02-06
- 修回日期: 2018-02-24
引用本文 |



2. 100021 北京,国家癌症中心,中国医学科学院北京协和医学院肿瘤医院;
3. 200433 上海,第二军医大学基础医学部生物化学与分子生物学教研室;
4. 100038 北京,首都医科大学附属北京世纪坛医院普外四科/临床营养科
2. National Cancer Center, Cancer Hospital, Chinese Academy of Medical Sciences and Peking Union Medical College, Beijing 100021, China;
3. Department of Biochemistry and Molecular Biology, College of Basic Medical Sciences, The Second Military Medical University, Shanghai 200433, China;
4. Department of Clinical Nutrition/General Surgery, Beijing Shijitan Hospital, Capital Medical University, Beijing 100038, China
二甲双胍是广泛应用于临床的口服降糖药物,能将糖尿病患者血糖降低3 mmol/L左右,并且对正常人的血糖无显著影响[1]。多项临床研究均证实二甲双胍能够抑制2型糖尿病(type 2 diabetes mellitus, T2DM)患者多种肿瘤的发生和发展,包括胰腺癌[2-3]、乳腺癌[4-5]、食管癌[6-7]、胃癌[8-9]、肝癌[10]、结直肠癌[11-12]、肺癌[13]、子宫内膜癌[14]和胶质母细胞瘤[15]等。
Warburg效应认为,正常细胞依靠线粒体氧化磷酸化产生ATP,肿瘤细胞则通过产能效率较低的有氧糖酵解为自身供能,但是糖酵解率比正常组织高200倍左右,因而肿瘤细胞对葡萄糖的依赖尤为明显。人体正常血糖为3.9~6.1 mmmol/L之间,在不同的营养状况下甚至可以降低至3.0 mmol/L以下。然而,目前细胞水平的肿瘤研究选用的培养基多含25 mmol/L的葡萄糖,远高于生理血糖水平,这种高糖环境为肿瘤细胞的生长增殖提供了最佳的外界条件[16],可能降低基础研究中药物抗肿瘤的疗效。因而,进行药物抗肿瘤作用研究时,降低机体血糖水平或许是一种可以尝试的辅助治疗方法。禁食以及能量限制的饮食方法均能够降低机体血糖水平并发挥一定的抗肿瘤作用[17-18]。最近有研究认为,体外研究使用的高糖培养基可能是二甲双胍无法发挥抗肿瘤作用的一种原因,当血糖水平降低时二甲双胍对肿瘤细胞的毒性不断增强[19-20]。
为了模拟人体正常血糖范围时的抗肿瘤作用,同时研究低糖能否增强二甲双胍对肿瘤细胞的杀伤作用,本研究选择不同葡萄糖浓度的培养基观察二甲双胍对肿瘤细胞的毒性,体内实验中,我们构建裸鼠皮下移植瘤模型,同时使用高脂低糖的生酮饮食(ketogenic diet, KD)降低裸鼠血糖水平以模拟体外的低糖环境。
1 材料与方法 1.1 细胞培养人胰腺癌细胞PANC-1、sw1990、AsPC-1、BxPC-3和人甲状腺癌细胞TPC-1、ARO、BCPAP、CAL-62均购自中国科学院上海细胞库,用DMEM培养基(含10%胎牛血清和100 u/ml的青链霉素,美国Gibco公司)培养于5%CO2、37℃培养箱中,隔天换培养液,胰蛋白酶消化传代。
1.2 细胞增殖检测2×104个细胞接种于96孔板中,细胞贴壁后随机分组,更换不同葡萄糖浓度(2.5、5、10、15、25 mmol/L)的培养基并加入不同浓度二甲双胍(0、2.5、5、10、15、20 mmol/L, Sigma-Aldrich),每组3个重复,培养1天后加入10 μl CCK-8溶液(上海同仁公司),并在培养箱中继续培养2 h后用酶标仪测定450 nm处的吸光度。
1.3 ATP检测细胞接种于6孔板培养24 h后吸去培养液,按增强型ATP检测试剂盒(上海碧云天)说明书操作计算相对ATP水平。
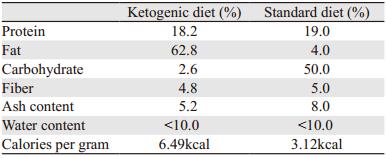
1.4 裸鼠皮下移植瘤模型建立及干预40只裸鼠适应性生长1周后,取对数生长期的胰腺癌细胞panc-1,用PBS调整细胞浓度为1×107/300 μl左右,于小鼠的右后支背部皮下接种细胞悬液300 μl。用游标卡尺测量肿瘤直径,按照V=ab2π/6(a为最大直径,b为最小直径)计算肿瘤体积,当肿瘤体积达100 mm3左右时,按移植瘤大小随机分成四组:正常饮食(standard diet, SD)组,SD+二甲双胍(metformin, Met)组,KD(深圳奇酮公司)组,KD+Met组,其中二甲双胍剂量为200 mg/kg,腹腔注射,每天一次,未用二甲双胍组注射等体积的0.9%氯化钠溶液。根据前期预实验结果,KD剂量为125 g/kg,SD不限量,小鼠每日摄取的总能量相近,SD及KD具体营养素配比见表 1。实验过程中使用雅培血糖仪尾静脉取血检测血糖水平。
使用SPSS19.0和Graphpad Prism 7.0软件进行统计学分析。计量资料数据用(x±s)表示,采用双尾t检验比较肿瘤体积和血糖水平,单因素方差分析比较组间差异,组间两两比较采用q检验,P < 0.05为差异有统计学意义。
2 结果 2.1 低糖增强胰腺癌细胞对二甲双胍的敏感度随着培养基中葡萄糖浓度的降低,二甲双胍显著抑制胰腺癌细胞PANC-1、sw1900、AsPC-1和BxPC-3的增殖,并且这四种细胞在2.5 mmol/L和5 mmol/L葡萄糖浓度下,二甲双胍浓度为5 mmol/L即可见显著抑制作用,见图 1。在高糖水平(15 mmol/L和25 mmol/L),二甲双胍浓度为15 mmol/L时,PANC-1细胞增殖才受到明显抑制,见图 1A。而sw1900、AsPC-1、BxPC-3细胞在二甲双胍浓度为20 mmol/L时才出现较为明显的抑制作用,见图 1B-1D。

|
| A: PANC-1; B: sw1990; C: AsPC-1; D: BxPC-3 图 1 低糖增加胰腺癌细胞对二甲双胍治疗的敏感度 Figure 1 Low glucose increased sensitivity of pancreatic cancer cells to metformin treatment |
葡萄糖浓度为2.5 mmol/L时,胰腺癌PANC-1、sw1990、AsPC-1和BxPC-3细胞在10 mmol/L二甲双胍中培养24 h后ATP水平显著下降(P < 0.01),见图 2A-2D。但是葡萄糖浓度为25 mmol/L时,PANC-1 ATP水平下降(P < 0.05),但较低糖组不明显,见图 2A。此外,高糖时二甲双胍对AsPC-1和BxPC-3细胞ATP水平并未产生显著影响,见图 2B-2D。

|
| A: PANC-1; B: sw1990; C: AsPC-1; D: BxPC-3; *: P < 0.05; **: P < 0.01; ns: no significance 图 2 二甲双胍降低低糖培养基中胰腺癌细胞ATP水平 Figure 2 Metformin decreased ATP levels in pancreatic cancer cells in medium containing low glucose |
与胰腺癌细胞类似,与相应对照组相比低糖(2.5 mmol/L葡萄糖)时,甲状腺癌ARO、TPC-1、BCPAP和CAL-62四种细胞存活率显著降低(P < 0.01),见图 3。而高糖(25 mmol/L葡萄糖)时,BCPAP存活率降低(P < 0.05),见图 3C,而ARO、TPC-1和CAL-63细胞存活率差异无统计学意义,见图 3A、3B和3D。

|
| A: ARO; B: TPC-1; C: BCPAP; D: CAL-62; *: P < 0.05; **: P < 0.01; ns: no significance 图 3 低糖增加甲状腺癌细胞对二甲双胍治疗的敏感度 Figure 3 Low glucose increased thyroid cancer cells to metformin treatment |
为模拟体内低糖环境,我们构建了人胰腺癌PANC-1荷瘤鼠模型并使用KD降低裸鼠血糖水平。裸鼠平均血糖为6 mmol/L左右,正常饮食加二甲双胍并未明显降低血糖水平,但是KD能够显著降低血糖(P < 0.01),并且KD喂养的裸鼠腹腔注射二甲双胍也未显著影响血糖水平,见图 4A。实验干预8周后,SD+Met组肿瘤抑制不明显,KD显著抑制肿瘤生长,且与KD组相比,KD+Met组进一步抑制移植瘤生长(P < 0.01),见图 4B-4D。

|
| SD: standard diet; Met: metformin; KD: ketogenic diet; *: P < 0.05; **: P < 0.01; ns: no significance 图 4 生酮饮食降低血糖水平,增强二甲双胍对PANC-1皮下移植瘤的抑制作用 Figure 4 Ketogenic diet reduced serum glucose concentration and enhanced metformin effects on reducing the growth of PANC-1 xenografts |
二甲双胍是T2DM一线治疗药物,近年来研究发现,它还存在潜在的抗肿瘤作用。早期的观察性研究显示,在使用二甲双胍的糖尿病患者中,肿瘤的发生风险显著降低,部分肿瘤的进展甚至被阻滞[21-22],动物实验同样发现二甲双胍对多种肿瘤生长的抑制作用。但是无论临床试验还是动物研究,血糖水平常在5~7 mmol/L左右,显著低于细胞研究中常用的25 mmol/L高糖培养基中的葡萄糖浓度。研究表明,高糖环境一方面为肿瘤提供最适的生长环境,另一方面能够掩盖二甲双胍在肿瘤治疗中对相关通路的调节作用。也正因此,很多细胞水平的研究发现正常浓度的二甲双胍对某些肿瘤细胞的增殖并未产生显著的抑制作用,只有在高浓度时才发挥一定的疗效[23-24]。
本研究发现,培养基中葡萄糖水平较高时(≥10 mmol/L)二甲双胍对胰腺癌和甲状腺癌细胞增殖抑制不明显,但是在低糖培养基(≤5 mmol/L)中,10 mmol/L二甲双胍即能明显抑制胰腺癌和甲状腺癌细胞增殖。有研究表明,二甲双胍能够抑制线粒体电子传递链复合体Ⅰ的功能从而影响胞内的氧化磷酸化,因此,细胞内的ATP水平会显著降低[25-26],本研究也检测了不同葡萄糖浓度下,二甲双胍干预1天后细胞内的ATP水平,结果发现,2.5 mmol/L葡萄糖浓度下,胰腺癌细胞内ATP水平显著降低,而25 mmol/L高糖培养基中细胞ATP水平虽有不同程度下降,但较低糖组不明显。许多研究者也发现了双胍类降糖药物与葡萄糖的这种协同作用[27-28]。Javeshghani等[29]观察到,结肠癌细胞对二甲双胍的敏感度依赖于供能物质的种类。当缺乏葡萄糖时,二甲双胍对结肠癌细胞的抑制作用明显增强,但是缺乏谷氨酰胺时无明显影响。Zhuang等[20]同样发现,低糖能够增强二甲双胍对乳腺癌细胞和卵巢癌细胞的毒性,降低胞内ATP水平。该研究还进一步阐明了低糖时二甲双胍抗肿瘤机制,他们发现二甲双胍能够显著降低磷酸化蛋白激酶B(phosphorylation-protein kinase B, p-Akt)和雷帕霉素靶蛋白(mammalian target of rapamycin, mTOR)的活性,并且这一作用不依赖单磷酸腺苷依赖的蛋白激酶(adenosine monophosphate activated protein kinase, AMPK)的激活。除此以外,二甲双胍还可通过激活诱导细胞周期停滞[30]、降低循环中胰岛素及胰岛素样生长因子水平[31]、抑制炎性反应及肿瘤内血管生成[32-33]、减少体内活性氧生成[34]等途径抑制肿瘤生长。
然而,我们可以在细胞水平将葡萄糖维持在极低浓度,但是正常机体血糖水平难以达到2.5 mmol/L。为此,我们选择高脂低糖的KD来模拟体外细胞实验的葡萄糖环境,结果发现KD喂养的小鼠血糖下降至3 mmol/L左右,并且KD联合二甲双胍组PANC-1移植瘤体积及重量显著小于单纯KD喂养组,而正常饮食下,二甲双胍对肿瘤无显著抑制作用。这一结果进一步证实了葡萄糖水平对二甲双胍抗肿瘤治疗敏感度的重要性,实验中我们也发现单纯KD即可显著抑制移植瘤生长,说明KD在其中也发挥一定的抗肿瘤作用,目前KD已知的抗肿瘤机制是多方面的[35],其产生的低糖环境,一方面减少肿瘤的葡萄糖来源,另一方面为机体正常细胞提供酮体作为能量来源,这为二甲双胍发挥抗肿瘤作用提供了最佳的外界环境。KD等显著降低血糖的饮食方式联合二甲双胍或许是一种切实可行的抗肿瘤治疗方案。接下来我们将进一步研究KD联合二甲双胍复杂的抗肿瘤机制,这对临床应用二甲双胍联合疗法抗肿瘤具有重要意义。
| [1] | Kodner C, Anderson L, Pohlgeers K. Glucose Management in Hospitalized Patients[J]. Am Fam Physician, 2017, 96(10): 648–54. |
| [2] | Kato K, Iwama H, Yamashita T, et al. The anti-diabetic drug metformin inhibits pancreatic cancer cell proliferation in vitro and in vivo: Study of the microRNAs associated with the antitumor effect of metformin[J]. Oncol Rep, 2016, 35(3): 1582–92. DOI:10.3892/or.2015.4496 |
| [3] | Duan W, Chen K, Jiang Z, et al. Desmoplasia suppression by metformin-mediated AMPK activation inhibits pancreatic cancer progression[J]. Cancer Lett, 2017, 385: 225–33. DOI:10.1016/j.canlet.2016.10.019 |
| [4] | Li NS, Zou JR, Lin H, et al. LKB1/AMPK inhibits TGF-beta1 production and the TGF-beta signaling pathway in breast cancer cells[J]. Tumour Biol, 2016, 37(6): 8249–58. DOI:10.1007/s13277-015-4639-9 |
| [5] | Wahdan-Alaswad R, Harrell JC, Fan Z, et al. Metformin attenuates transforming growth factor beta (TGF-beta) mediated oncogenesis in mesenchymal stem-like/claudin-low triple negative breast cancer[J]. Cell Cycle, 2016, 15(8): 1046–59. DOI:10.1080/15384101.2016.1152432 |
| [6] | Cai X, Hu X, Tan X, et al. Metformin Induced AMPK Activation, G0/G1 Phase Cell Cycle Arrest and the Inhibition of Growth of Esophageal Squamous Cell Carcinomas In Vitro and In Vivo[J]. PLoS One, 2015, 10(7): e0133349. DOI:10.1371/journal.pone.0133349 |
| [7] | Fujihara S, Kato K, Morishita A, et al. Antidiabetic drug metformin inhibits esophageal adenocarcinoma cell proliferation in vitro and in vivo[J]. Int J Oncol, 2015, 46(5): 2172–80. DOI:10.3892/ijo.2015.2903 |
| [8] | Greenhill C. Metformin improves survival and recurrence rate in patients with diabetes and gastric cancer[J]. Nat Rev Gastroenterol Hepatol, 2015, 12(3): 124. |
| [9] | Chen G, Feng W, Zhang S, et al. Metformin inhibits gastric cancer via the inhibition of HIF1alpha/PKM2 signaling[J]. Am J Cancer Res, 2015, 5(4): 1423–34. |
| [10] | Obara A, Fujita Y, Abudukadier A, et al. DEPTOR-related mTOR suppression is involved in metformin's anti-cancer action in human liver cancer cells[J]. Biochem Biophys Res Commun, 2015, 460(4): 1047–52. DOI:10.1016/j.bbrc.2015.03.148 |
| [11] | Abdelsatir AA, Husain NE, Hassan AT, et al. Potential Benefit of Metformin as Treatment for Colon Cancer: the Evidence so Far[J]. Asia Pac J Cancer Prev, 2015, 16(18): 8053–8. |
| [12] | He X-K, Su T, Si J, et al. Metformin Is Associated With Slightly Reduced Risk of Colorectal Cancer and Moderate Survival Benefits in Diabetes Mellitus: A Meta-Analysis[J]. Medicine (Baltimore), 2016, 95(7): e2749. DOI:10.1097/MD.0000000000002749 |
| [13] | Guo Q, Liu Z, Jiang L, et al. Metformin inhibits growth of human non-small cell lung cancer cells via liver kinase B-1-independent activation of adenosine monophosphate-activated protein kinase[J]. Mol Med Rep, 2016, 13(3): 2590–6. DOI:10.3892/mmr.2016.4830 |
| [14] | Wallbillich JJ, Josyula S, Saini U, et al. High Glucose-Mediated STAT3 Activation in Endometrial Cancer Is Inhibited by Metformin: Therapeutic Implications for Endometrial Cancer[J]. PLoS One, 2017, 12(1): e0170318. DOI:10.1371/journal.pone.0170318 |
| [15] | Carmignani M, Volpe AR, Aldea M, et al. Glioblastoma stem cells: a new target for metformin and arsenic trioxide[J]. J Biol Regul Homelst Agents, 2014, 28(1): 1–15. |
| [16] | Han L, Ma Q, Li J, et al. High glucose promotes pancreatic cancer cell proliferation via the induction of EGF expression and transactivation of EGFR[J]. PLoS One, 2011, 6(11): e27074. DOI:10.1371/journal.pone.0027074 |
| [17] | De Lorenzo MS, Baljinnyam E, Vatner DE, et al. Caloric restriction reduces growth of mammary tumors and metastases[J]. Carcinogenesis, 2011, 32(9): 1381–7. DOI:10.1093/carcin/bgr107 |
| [18] | Lee C, Longo VD. Fasting vs dietary restriction in cellular protection and cancer treatment: from model organisms to patients[J]. Oncogene, 2011, 30(30): 3305–16. DOI:10.1038/onc.2011.91 |
| [19] | Khurana A, Shridhar V. Shridhar. Metformin is synthetically lethal with glucose withdrawal in cancer cells[J]. Cell Cycle, 2012, 11(15): 2779–80. DOI:10.4161/cc.21394 |
| [20] | Zhuang Y, Chan DK, Haugrud AB, et al. Mechanisms by which low glucose enhances the cytotoxicity of metformin to cancer cells both in vitro and in vivo[J]. PLoS One, 2014, 9(9): e108444. DOI:10.1371/journal.pone.0108444 |
| [21] | Evans JM, Donnelly LA, Emslie-Smith AM, et al. Metformin and reduced risk of cancer in diabetic patients[J]. BMJ, 2005, 330(7503): 1304–5. DOI:10.1136/bmj.38415.708634.F7 |
| [22] | Ma S, Zheng Y, Xiao Y, et al. Meta-analysis of studies using metformin as a reducer for liver cancer risk in diabetic patients[J]. Medicine(Baltimore), 2017, 96(19): e6888. |
| [23] | Zhuang Y, Miskimins WK. Cell cycle arrest in Metformin treated breast cancer cells involves activation of AMPK, downregulation of cyclin D1, and requires p27Kip1 or p21Cip1[J]. J Mol Signal, 2008, 3: 18. DOI:10.1186/1750-2187-3-18 |
| [24] | Zhu Z, Jiang W, Thompson MD, et al. Metformin as an energy restriction mimetic agent for breast cancer prevention[J]. J Carcinog, 2011, 10: 17. DOI:10.4103/1477-3163.83043 |
| [25] | Cameron AR, Logie L, Patel K, et al. Metformin selectively targets redox control of complex Ⅰ energy transduction[J]. Redox Biol, 2018, 14: 187–97. DOI:10.1016/j.redox.2017.08.018 |
| [26] | Kalyanaraman B, Cheng G, Hardy M, et al. Mitochondria-targeted metformins: anti-tumour and redox signalling mechanisms[J]. Interface Focus, 2017, 7(2): 20160109. DOI:10.1098/rsfs.2016.0109 |
| [27] | Matsuo J, Tsukumo Y, Saito S, et al. Hyperactivation of 4E-binding protein 1 as a mediator of biguanide-induced cytotoxicity during glucose deprivation[J]. Mol Cancer Ther, 2012, 11(5): 1082–91. DOI:10.1158/1535-7163.MCT-11-0871 |
| [28] | Ben Sahra I, Laurent K, Giuliano S, et al. Targeting cancer cell metabolism: the combination of metformin and 2-deoxyglucose induces p53-dependent apoptosis in prostate cancer cells[J]. Cancer Res, 2010, 70(6): 2465–75. DOI:10.1158/0008-5472.CAN-09-2782 |
| [29] | Javeshghani S, Zakikhani M, Austin S, et al. Carbon source and myc expression influence the antiproliferative actions of metformin[J]. Cancer Res, 2012, 72(23): 6257–67. DOI:10.1158/0008-5472.CAN-12-2907 |
| [30] | Skinner HD, Crane CH, Garrett CR, et al. Metformin use and improved response to therapy in rectal cancer[J]. Cancer Med, 2013, 2(1): 99–107. DOI:10.1002/cam4.54 |
| [31] | Wang CF, Zhang G, Zhao LJ, et al. Overexpression of the insulin receptor isoform A promotes endometrial carcinoma cell growth[J]. PLoS One, 2013, 8(8): e69001. DOI:10.1371/journal.pone.0069001 |
| [32] | Wang J, Li G, Wang Y, Tang S, et al. Suppression of tumor angiogenesis by metformin treatment via a mechanism linked to targeting of HER2/HIF-1alpha/VEGF secretion axis[J]. Oncotarget, 2015, 6(42): 44579–92. |
| [33] | Tadakawa M, Takeda T, Li B, et al. The anti-diabetic drug metformin inhibits vascular endothelial growth factor expression via the mammalian target of rapamycin complex 1/hypoxia-inducible factor-1alpha signaling pathway in ELT-3 cells[J]. Mol Cell Endocrinol, 2015, 399: 1–8. DOI:10.1016/j.mce.2014.08.012 |
| [34] | Khouri H, Collin F, Bonnefont-Rousselot D, et al. Radical-induced oxidation of metformin[J]. Eur J Biochem, 2004, 271(23-24): 4745–52. DOI:10.1111/ejb.2004.271.issue-23-24 |
| [35] | Boison D. New insights into the mechanisms of the ketogenic diet[J]. Curr Opin Neurol, 2017, 30(2): 187–92. DOI:10.1097/WCO.0000000000000432 |