 2018, Vol. 45
2018, Vol. 45文章信息
- 乳腺癌基因分型与患者临床病理及预后的关系
- Correlation of Genotypes with Clinicopathological and Prognostic Characteristics of Breast Cancer Patients
- 肿瘤防治研究, 2018, 45(10): 752-757
- Cancer Research on Prevention and Treatment, 2018, 45(10): 752-757
- http://www.zlfzyj.com/CN/10.3971/j.issn.1000-8578.2018.17.1658
- 收稿日期: 2017-12-26
- 修回日期: 2018-06-21
引用本文 |



2. 524002 湛江,广东省农垦中心医院检验科;
3. 529030 江门,江门市妇幼保健院药学部;
4. 529030 江门,江门市中心医院病理科
2. Laboratory Department, Central Hospital of Guangdong Nongken, Zhanjiang 524002, China;
3. Pharmacy Department, Jiangmen Maternity and Child Health Care Hospital, Jiangmen 529030, China;
4. Pathology Department, Jiangmen Central Hospital, Jiangmen 529030, China
乳腺癌(Breast cancer)是最常见的女性肿瘤,每年全球新增女性患者约为167.7万例(发病率为52.7/10万人),导致的死亡病例约为52.2万例(死亡率为13.2/10万人)[1]。虽然我国乳腺癌的患病率(37.9/10万人)和死亡率(9.2/10万人)均低于世界平均水平[2],但多项流行病学调查显示,我国乳腺癌发病率有上升趋势[3-4],严重威胁我国妇女健康。临床上,雌激素受体(estrogen receptor, ER)、孕激素受体(progesterone receptor, PR)及人表皮生长因子受体2(human epidermal growth factor receptor 2, HER2)为代表的免疫组织化学表型分类已得到广泛运用,大致分为管腔癌型(luminal,ER阳性为特征)、HER2过表达型(HER2+,ER阴性、HER2阳性或扩增)和三阴性乳腺癌(triple negative breast cancer, TNBC)三大类[5]。虽然免疫组织化学分型对临床治疗策略选择有重要意义,但是同样的免疫组织化学亚型可能有着各异的临床预后及药物反应特点。
本世纪初高通量检测方法的兴起,为系统研究肿瘤异常分子调控网络提供了手段,其中最具开创性的研究是,Perou等[6]将乳腺癌按表达特征进行亚型分类,开启了肿瘤基因分型研究的时代。为了开发能在临床全面推广的乳腺癌基因分型方法,近年来研究者从不同的技术平台上研究出多种方法[7-9]。为了评价以绝对表达量为分型判断依据的新型乳腺癌分型程序AIMS(absolute intrinsic molecular subtyping)的适用性,我们对ArrayExpress数据库和TCGA公开的乳腺癌表达谱数据进行分型处理,并通过分析AIMS基因分型和患者临床病理及预后特征的相关性,初步判断AIMS的临床应用价值,为乳腺癌治疗策略及预后随访的精准化提供有力的指导。
1 材料与方法 1.1 Array Express数据库(AE-meta)乳腺癌表达谱检索与下载利用Array Express数据库(http://www.ebi.ac.uk/arrayexpress/),搜索并收录了53个原始数据和临床数据较完整的,使用Affymetrix U133 Plus 2.0芯片的乳腺癌相关数据集,包括:E-GEOD-10780、E-GEOD-10810、E-GEOD-11001、E-GEOD-12276、E-GEOD-16391、E-GEOD-16446、E-GEOD-17907、E-GEOD-18864、E-GEOD-20685、E-GEOD-20711、E-GEOD-21422、E-GEOD-21653、E-GEOD-22035、E-GEOD-22513、E-GEOD-22544、E-GEOD-23177、E-GEOD-23593、E-GEOD-23720、E-GEOD-25407、E-GEOD-26457、E-GEOD-28796、E-GEOD-28844、E-GEOD-29431、E-GEOD-30010、E-GEOD-32646、E-GEOD-36771、E-GEOD-3744、E-GEOD-38554、E-GEOD-42568、E-GEOD-43358、E-GEOD-43365、E-GEOD-43502、E-GEOD-46222、E-GEOD-47109、E-GEOD-47389、E-GEOD-48390、E-GEOD-50567、E-GEOD-54002、E-GEOD-5460、E-GEOD-58792、E-GEOD-58812、E-GEOD-61304、E-GEOD-65194、E-GEOD-71258、E-GEOD-76275、E-GEOD-7904、E-GEOD-9195、E-MTAB-1006、E-MTAB-2501、E-MTAB-365、E-MTAB-748、E-TABM-276、E-TABM-854。
1.2 利用Affy和YuGene程序对表达谱进行整合分析在R语言(版本3.1.3)程序平台上,安装Affy程序(版本1.46.1)处理每个表达谱数据;在R语言(版本3.2.2)程序平台上,安装YuGene程序(版本1.1.4)处理每个表达谱数据。参考文献[10],利用模拟代码1用于芯片的背景校正;再利用模拟代码2用于将同一基因的多个探针合并,选用最高平均表达值的探针代表该基因;最后利用模拟代码3对每个样品进行归一化,得到每个基因的YuGene值(Y-score)。整合ArrayExpress数据库53个乳腺癌相关表达谱,得到乳腺正常组织522例,乳腺癌组织5 199例,并命名为AE-meta乳腺癌数据集。
模拟代码:
(1)R>expresso(X, bg.correct=TRUE, bgcorrect.method="rma", normalize=FALSE, pmcorrect.method="pmonly", summary.method="avgdiff")
(2)R>X[which(!duplicated(X$Gene_ID)), ]
(3)R>YuGene(X)
1.3 TCGA乳腺癌表达谱数据集下载下载癌症基因组图谱(The Cancer Genome Atlas, TCGA)中浸润性乳腺癌(Breast invasive carcinoma, BRCA)的RNA测序数据,利用Excel 2010合并整理,再用YuGene进行归一化处理,得到113例癌旁正常对照组织,1 095例乳腺癌组织的TCGA乳腺癌数据集。
1.4 用AIMS程序包进行分析在R语言(版本3.2.2)程序平台上,安装AIMS程序包(版本1.2.0)处理每个表达谱数据集。参考文献[11],利用模拟代码4用于对每一个乳腺癌患者进行基因分型,包括管腔癌A型(Luminal A, LumA)、管腔癌B型(Luminal B, LumB)、人表皮生长因子受体2过表达型(HER2 overexpression, HER2)、基底细胞样型(Basal-like, Basal)和正常细胞样型(Normal-like, NorL)。
模拟代码:(4)R>applyAIMS (X, Gene_ID)
1.5 统计学方法采用Excel2010, GraphPad5.0软件进行数据处理,其中AIMS分型与临床指标的相关性用卡方检验,生存分析使用Kaplan-Meier法、Log rank检验,检验水平α=0.05。
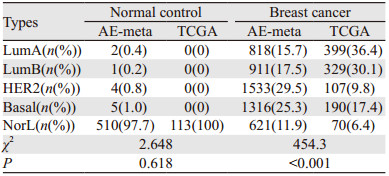
2 结果 2.1 AE-meta和TCGA数据集中各基因亚型的分布比例乳腺正常对照组织绝大部分被归为NorL型,两数据集间差异无统计学意义(P > 0.05)。乳腺癌组织中,AE-meta和TCGA的分布差异较大(P < 0.05),其中AE-met占有较多的HER2型和Basal型,而TCGA中LumA型和LumB型较多,见表 1。
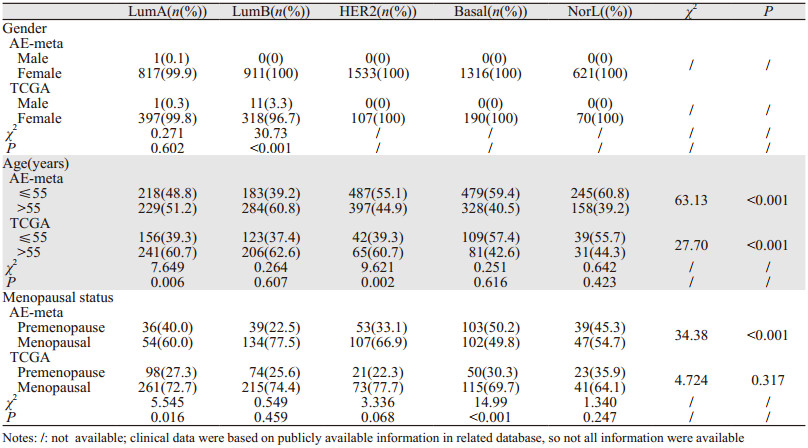
在患者一般信息中,各基因亚型的年龄分布差异有统计学意义(P < 0.05),LumA和LumB型中,患者年龄 > 55岁的占比大,而Basal和NorL型中,患者年龄≤55岁的比例较大;此外,基因分型间性别及月经状况的分布未见规律,见表 2。在患者临床病理特征中,LumA和NorL型中高级别的病理分级较少,而Basal型中高级别的病理分级较多;在T分期中,LumA型T1分期的比例较大,Basal型T2分期的比例较大;在N分期中,HER2型N2-3分期的比例较大,LumA和Basal型N0分期的比例较大;而M分期及总的临床分期中,基因分型间分布未见规律,见表 3。
|

|
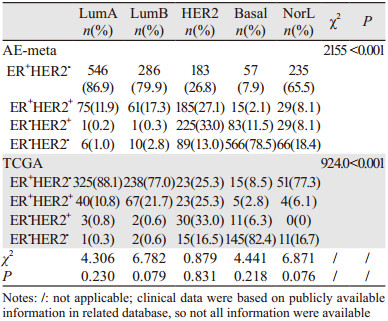
在ER及HER2的免疫组织化学表达状态中,各基因亚型间受体表达阳性率差异有统计学意义(P < 0.05),而AE-meta数据集与TCGA数据集间,受体表达阳性率分布有较好的一致性(P > 0.05),见表 4。LumA型中 > 85%为ER+HER2-类、10%为ER+HER2+类;LumB型中 > 75%为ER+HER2-类、20%为ER+HER2+类;NorL型中约70%为ER+HER2-类,17%为ER-HER2-类;Basal型中80%为ER-HER2-类;HER2型中33%为ER-HER2+类,26%为ER+HER2+类。
|
整合AE-meta数据集和TCGA数据集中全部的预后信息,在总生存时间曲线中,LumA型的生存预后最好,其次是NorL型,而Basal、HER2和LumB型的生存预后最差,三者曲线有相交;其中Basal和HER2型生存曲线的下降斜率成先高后低的趋势,而LumB型生存曲线的下降斜率则基本不变,见图 1A。在无复发生存时间曲线中,LumA型的复发最少,其次是NorL和LumB型,而Basal和HER2型复发最多最快,见图 1B。在无局部复发生存时间曲线中,各基因亚型间局部复发情况均较少,且曲线交错,见图 1C。在无远处转移生存生存曲线中,LumA型的远处转移率最小,其次是NorL和LumB型,而Basal和HER2型远处转移最多最快,见图 1D。

|
| 图 1 各基因分型间乳腺癌患者的生存曲线 Figure 1 Survival curves of breast cancer patients with different genotypes |
在无骨转移生存时间曲线中,各基因亚型间骨转移情况均较多,且曲线交错,见图 2A。在无脑转移生存时间曲线中,Basal型的脑转移最快最多,而其他亚型的脑转移情况均较少,见图 2B。在无肝转移生存时间曲线中,LumA和NorL型的肝转移最少,Basal和HER2型的肝转移最快最多,见图 2C。在无肺转移生存时间曲线中,各基因亚型见肺转移情况均较多,且曲线交错,见图 2D。在各基因亚型的转移灶分布情况中,骨转移占比最小的是Basal型,最多的是NorL型;脑转移占比最多的是Basal型;肝转移占比较多的是HER2和Basal型;肺转移占比最多的是LumB型,见图 2E和2F。

|
| 图 2 各基因分型乳腺癌患者的骨、脑、肝和肺转移情况 Figure 2 Bone, brain, liver and lung metastasis of breast cancer patients with different genotypes |
肿瘤基因分型研究始于Perou等[6]的研究,而AIMS的模型也源于该研究鉴定出的5大乳腺癌基因亚型:LumA、LumB、HER2、Basal和NorL;其中LumA、LumB和NorL三个分子亚型一般为ER+乳腺癌,HER2型则大部分为HER2+的乳腺癌,而Basal型包含了大量TNBC[6, 11-13]。本课题组观察到两个独立数据集(AE-meta和TCGA),正常乳腺对照组织基本(> 97%)被定义为NorL型,被定义为LumA和LumB型的乳腺癌样品基本(> 96%)为ER+,被定义为HER2型的乳腺癌样品中大部分(> 58%)为HER2+,被定义为Basal型的乳腺癌样品大部分(> 78%)为ER-HER2-。两个数据集中,各基因亚型比例差异较大,其原因主要在于,TCGA的乳腺癌样品是非定向收集的,样品组成的与临床乳腺癌各亚型分布较一致[6](LumA: 36.4%, LumB: 30.1%, HER2: 9.8%, Basal: 17.4%, NorL: 6.4%);而整合的AE-meta数据集中,有多个实验数据是针对HER2+乳腺癌或TNBC,因此,AE-meta数据集中HER2和Basal型的比例较高(29.5%和25.3%),表明AIMS程序得到的乳腺癌基因分型结果有较好的稳定性,并与受体免疫组织化学表达情况有较好的一致性。
生存预后分析中,本研究观察到LumA型的复发转移最少及生存情况最佳,Basal和HER2型的复发转移最多且生存情况最差,与大量的回顾性研究结果相吻合[11, 13-14]。本研究观察到Basal和HER2型的总生存及无复发生存曲线均有较明显的平台效应,两亚型生存曲线分别在前20月和40月呈陡直下降趋势,而在60月和80月后呈缓慢下降趋势;而LumB型的总生存及无复发生存曲线则呈匀速下降趋势,在100月及120月附近逼近甚至越过Basal和HER2型的生存时间曲线,提示,针对Basal和HER2型要防控治疗后的迅速耐药复发进展,而针对LumB型则要在长时间内保持较高频率的复查,但其背后的生物学及分子机制还有待深入探讨。
在转移靶器官的选择性分析中,本研究观察到各基因亚型对骨及肺都有着较为强烈的转移倾向,且各亚型间风险差异并不显著,在脑转移中,Basal型的风险较高,在肝转移中,Basal和HER2型的风险较高。虽然有文献报道[14-15],Basal型的内脏转移风险较高,骨转移风险较低,但我们研究发现,Basal型对四大靶器官的转移风险都较高,不应该忽视Basal型的骨转移情况。
在众多乳腺癌基因分型方法中,美国国立综合癌症网络(National Comprehensive Cancer Network)推荐的乳腺癌DX21基因检测(Oncotype DX 21 breast cancer assays)和获得美国食品药品监督管理局(Food and Drug Administration)批准用于临床的PAM50基因检测(PAM50-based Prosigna risk of recurrence score)最为著名。虽然国外大量研究已证实,包括DX21和PAM50在内的乳腺癌基因检测分型技术能指导临床用药及复发风险预判[7],但上述的两种方法未在中国市场进行大规模验证。此外,上述基因分型过程中需要进行样品间的归一化[16-17],导致分型结果与收集样品量多少相关,使得开发上述两种分型方法的公司能控制最核心的数据,形成技术壁垒。本研究选用的AIMS乳腺癌分型程序,最大优势是不需要进行样品间的表达量校正,只需在同一样品内比较不同基因的相对表达量,能在各种检测平台中得到稳定的分型结果[11]。
综上所述,AMIS程序得到的乳腺癌分型能有效反映患者临床病理特征,为患者的预后风险判断提供帮助,具有指导临床诊治的潜在价值,值得我们进一步的临床实验验证。
| [1] | Torre LA, Bray F, Siegel RL, et al. Global cancer statistics, 2012[J]. CA Cancer J Clin, 2015, 65(2): 87–108. DOI:10.3322/caac.21262 |
| [2] | Zheng R, Zeng H, Zhang S, et al. National estimates of cancer prevalence in China, 2011[J]. Cancer Lett, 2016, 370(1): 33–8. DOI:10.1016/j.canlet.2015.10.003 |
| [3] | 陈万青, 郑荣寿. 中国女性乳腺癌发病死亡和生存状况[J]. 中国肿瘤临床, 2015, 42(13): 668–74. [ Chen WQ, Zheng RS. Incidence, mortality and survival analysis of breast cancer in China[J]. Zhongguo Zhong Liu Lin Chuang, 2015, 42(13): 668–74. ] |
| [4] | Chen W, Zheng R, Zeng H, et al. Annual report on status of cancer in China, 2011[J]. Chin J Cancer Res, 2015, 27(1): 2–12. |
| [5] | Costa A, Kieffer Y, Scholer-Dahirel A, et al. Fibroblast Heterogeneity and Immunosuppressive Environment in Human Breast Cancer[J]. Cancer Cell, 2018, 33(3): 463–79. DOI:10.1016/j.ccell.2018.01.011 |
| [6] | Perou CM, Sørlie T, Eisen MB, et al. Molecular portraits of human breast tumours[J]. Nature, 2000, 406(6797): 747–52. DOI:10.1038/35021093 |
| [7] | Harbeck N, Sotlar K, Wuerstlein R, et al. Molecular and protein markers for clinical decision making in breast cancer: today and tomorrow[J]. Cancer Treat Rev, 2014, 40(3): 434–44. DOI:10.1016/j.ctrv.2013.09.014 |
| [8] | Guedj M, Marisa L, de Reynies A, et al. A refined molecular taxonomy of breast cancer[J]. Oncogene, 2012, 31(9): 1196–206. DOI:10.1038/onc.2011.301 |
| [9] | Lehmann BD, Bauer JA, Chen X, et al. Identification of human triple-negative breast cancer subtypes and preclinical models for selection of targeted therapies[J]. J Clin Invest, 2011, 121(7): 2750–67. DOI:10.1172/JCI45014 |
| [10] | Lê Cao KA, Rohart F, McHugh L, et al. YuGene: a simple approach to scale gene expression data derived from different platforms for integrated analyses[J]. Genomics, 2014, 103(4): 239–51. DOI:10.1016/j.ygeno.2014.03.001 |
| [11] | Paquet ER, Hallett MT. Absolute assignment of breast cancer intrinsic molecular subtype[J]. J Natl Cancer Inst, 2015, 107(1): 357. |
| [12] | Goldhirsch A, Wood WC, Coates AS, et al. Strategies for subtypes-dealing with the diversity of breast cancer: highlights of the St. Gallen International Expert Consensus on the Primary Therapy of Early Breast Cancer 2011[J]. Ann Oncol, 2011, 22(8): 1736–47. DOI:10.1093/annonc/mdr304 |
| [13] | Sørlie T, Perou CM, Tibshirani R, et al. Gene expression patterns of breast carcinomas distinguish tumor subclasses with clinical implications[J]. Proc Natl Acad Sci U S A, 2001, 98(19): 10869–74. DOI:10.1073/pnas.191367098 |
| [14] | Kennecke H, Yerushalmi R, Woods R, et al. Metastatic behavior of breast cancer subtype[J]. J Clin Oncol, 2010, 28(20): 3271–7. DOI:10.1200/JCO.2009.25.9820 |
| [15] | Dent R, Trudeau M, Pritchard KI, et al. Triple-negative breast cancer: clinical features and patterns of recurrence[J]. Clin Cancer Res, 2007, 13(15 Pt 1): 4429–34. |
| [16] | Lusa L, McShane LM, Reid JF, et al. Challenges in projecting clustering results across gene expression-profiling datasets[J]. J Natl Cancer Inst, 2007, 99(22): 1715–23. DOI:10.1093/jnci/djm216 |
| [17] | Weigelt B, Mackay A, A' hern R, et al. Breast cancer molecular profiling with single sample predictors: a retrospective analysis[J]. Lancet Oncol, 2010, 11(4): 339–49. DOI:10.1016/S1470-2045(10)70008-5 |