 2018, Vol. 45
2018, Vol. 45文章信息
- 皮下脂膜炎样T细胞淋巴瘤的临床特征及预后分析
- Clinical Features and Prognosis of Subcutaneous Panniculitis-like T-cell Lymphoma
- 肿瘤防治研究, 2018, 45(7): 475-478
- Cancer Research on Prevention and Treatment, 2018, 45(7): 475-478
- http://www.zlfzyj.com/CN/10.3971/j.issn.1000-8578.2018.17.1329
- 收稿日期: 2017-10-23
- 修回日期: 2018-02-24
引用本文 |



皮下脂膜炎样T细胞淋巴瘤(subcutaneous panniculitis-like T-cell lymphoma, SPTCL)是一种罕见的细胞毒性T细胞淋巴瘤,占所有非霍奇金淋巴瘤的1%以下[1],占原发皮肤的T细胞淋巴瘤的1%,主要累及皮下脂肪组织,部分患者有骨髓侵犯及伴发噬血细胞综合征(hemophagocytic syndrome, HPS),后者病情进展迅速、预后差[2],国内尚无大样本资料报道。本研究回顾性分析北京大学第三医院血液科5例临床资料,探讨SPTCL的临床及病理特点。
1 资料与方法 1.1 临床资料2005年1月至2016年12月在本院确诊的皮下脂膜炎样T细胞淋巴瘤患者共5例,所有患者病例资料完整,接受了规律治疗。5例患者中男3例,女2例,平均发病年龄为35岁,中位年龄32(15~73)岁。5例患者均有皮下结节的临床表现,中位发病时间(发病到确诊时间)为3(1~36)月,发病时2例伴发热,其中1例同时伴有体质量下降及盗汗。
1.2 临床诊断及分期 1.2.1 影像学检查全身淋巴结彩超、CT、MRI、PET/CT等。按照Ann Arbor分期及皮肤淋巴瘤的TNM分期方案进行临床分期,乳酸脱氢酶(LDH)及β2微球蛋白(β2-MG)等检查评估预后,见表 1。
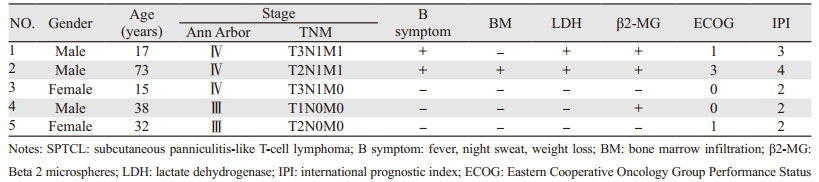
应用美国东部肿瘤协作组(Eastern Cooperative Oncology Group, ECOG)评分进行体力状况评估:0分:活动能力完全正常,与起病前活动能力无任何差异;1分:能自由走动及从事轻体力活动,包括一般家务或办公室工作,但不能从事较重的体力活动;2分:能自由走动及生活自理,但已丧失工作能力,日间不少于一半时间可以起床活动;3分:生活仅能部分自理,日间一半以上时间卧床或坐轮椅;4分:卧床不起,生活不能自理。
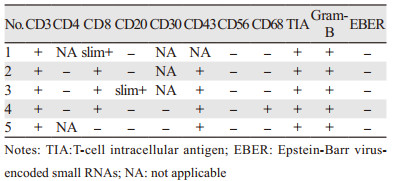
1.3 病理诊断免疫组织化学抗体包括CD3、CD4、CD8、CD56、CD68、CD20、Ki-67、TIA、Gram-B、EBER等,见表 2。全部患者的病理报告均由病理科医师复诊后确诊作出诊断,诊断标准详见2016年WHO诊断标准[3]。
|
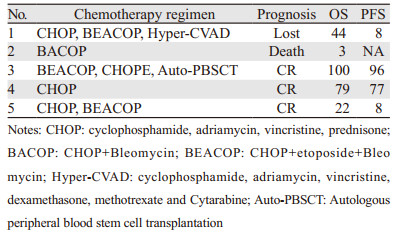
5例患者均接受了多药联合化疗,初始治疗中3例应用CHOP(环磷酰胺、阿霉素、长春新碱、泼尼松)方案,2例应用BEACOP方案(CHOP联合依托泊苷+博莱霉素);5例中的1例接受自体干细胞移植,见表 3。
疗效评价标准参照淋巴瘤国际协调组织2007年制订的恶性淋巴瘤疗效评价标准[4]:完全缓解(complete remission, CR)、部分缓解(partial remission, PR)、稳定(stable disease, SD)及进展(progression disease, PD)。
1.6 随访门诊及电话随访至2017年10月,死亡及失访病例以死亡、失访时间为随访终点。确诊至复发时间为无进展生存(progression-free survival, PFS)时间,确诊至死亡时间或随访终点为总生存(overall survival, OS)时间。
1.7 统计学方法应用SPSS19.0软件对数据进行统计学描述,包括应用Means过程进行均值、中位值的计算及OS和PFS的计算。
2 结果 2.1 临床分期5例患者均为临床晚期患者,其中3例患者Ⅲ期,2例患者Ⅳ期。
2.2 实验室检查2例患者血清乳酸脱氢酶(lactate dehydrogenase, LDH)升高,3例患者血清β2微球蛋白(Beta 2 microspheres, β2-MG)升高。1例骨髓活检证实存在骨髓累及,余4例阴性。
2.3 病理特点4例患者表现为皮下组织较少,可见脂肪小叶内淋巴细胞弥漫性增生浸润,2例患者脂肪组织内可见多量上皮样结节,增生的细胞体积中等偏大,核仁明显,1例患者核碎明显,伴有吞噬现象。5例患者均为CD3+,TIA+,Gram-B+,EBER-,见表 2。
2.4 治疗及预后初始方案治疗后2例完全缓解,2例部分缓解,1例未缓解。随访截至2017年10月,中位随访时间44(3~100)月,5例患者中存活3例,见表 3。
3 讨论SPTCL临床上非常罕见,病因及发病机制尚不清楚,过去常将其误诊为良性脂膜炎、湿疹、银屑病及蜂窝组织炎等其他疾病[5]。1991年Gonzalez等[6]第一次提出这一概念为α/β和γ/δ表型的脂膜炎样T细胞淋巴瘤,2005年WHO将其正式定义为原发于皮下的细胞毒性T细胞淋巴瘤[7],2016年WHO将其归类于成熟T细胞和NK细胞肿瘤中的一种[3]。本病好发于青壮年,主要表现为非特异性皮下结节或红斑,四肢最常见,尤以双下肢为著,其次为躯干,发病病因及机制不清,约20%的患者可伴有噬血细胞综合征,为SPTCL的主要死亡原因之一[8]。临床上单纯的SPTCL预后较好,全身受累或继发噬血细胞综合征者病情进展迅速,预后差。鉴别诊断主要针对以下几种疾病:(1)非淋巴瘤性脂膜炎:组织学可见多量脂肪细胞坏死,中性粒细胞、组织细胞浸润多见,有泡沫细胞形成,免疫表型为多克隆性,主要为B淋巴细胞、组织细胞和浆细胞,预后较好;(2)组织细胞吞噬性脂膜炎(CHP):结节质地较软,病程较缓,可达数年之久。CHP早期至终末期均没有发现淋巴瘤细胞的证据,也没有EBV感染相关证据,对免疫抑制剂如环孢素、泼尼松的治疗反应也较好;(3)原发性皮肤CD30+间变性大细胞淋巴瘤(CD30+ALCL):好发于中老年,预后较好。临床表现为表浅的红斑或皮肤结节,局限于真皮,脂肪组织受累较轻。CD30+大细胞在皮肤活检标本中占75%以上或大团簇集,而SPTCL表达CD30+细胞较少用以鉴别。
本组5例患者均以结节、肿块起病,且均为多发,2例伴脾大、1例累及骨髓、1例累及乳腺、1例累及口咽;5例患者均为中晚期患者,3例无B症状者预后较好,另外2例中1例仅有发热者PFS为8月,截止到失访时生存44月,另1例发病即出现所有B症状,其后继发噬血细胞综合征,病情进展迅速,仅生存3月。5例患者采用了两种分期方法,从临床结果分析TNM分期中不存在内脏侵犯的3例患者均存活,可能对临床有一定的预后提示作用。
SCPTL的确诊依靠典型的临床表现和病理诊断,其组织学特征类似于脂膜炎,2008年Willemze等[8]总结了63例SPTCL患者的病理结果,主要病理特征如下:大小不一的非典型性淋巴细胞浸润皮下脂肪组织,并围绕单个脂肪细胞呈花边样排列,常见核分裂、脂肪组织坏死、组织细胞吞噬现象及凋亡小体,并可伴有血管浸润和肉芽肿样改变,免疫表型常为CD3、CD8阳性,CD4、CD56阴性,TCRβF1阳性,表达细胞毒颗粒相关蛋白(TIA-1、Gram-B、perforin),细胞增殖指数常 > 25%。本组5例患者CD3、TIA-1、Gram-B均为阳性,4例CD8阳性,4例患者进行了CD43的检测均为阳性,3例患者检测了CD4,5例患者的CD56和EBER均为阴性,1例患者CD68阳性,1例患者CD20散在阳性。
目前SCPTL的治疗尚无明确共识,Willemze建议对于不伴有HPS的患者,可将免疫抑制治疗作为一线治疗,CHOP样方案化疗的完全缓解率可达到64%[8],其他报道也指出免疫抑制剂(如环孢素等)在治疗不伴有HPS的SPTCL患者可取得较好效果[9-11]。本研究中3例患者初始治疗为CHOP方案,其中仅有1例2周期后达到CR,完成6周期CHOP方案后予亚砷酸维持3周期,至今CR,其PFS为77月;1例在3周期CHOP后达到PR,更换为BEACOP方案后2周期达CR,因严重骨髓抑制改为CHOP方案后复发,其后更换为BEACOP方案2周期后再次达CR,后予MTX及6-MP维持治疗3月,发病至今22月仍为CR;1例在2周期CHOP后达到PR,更换为BEACOP方案后2周期达CR,后予Hyper-CVAD强化2周期,因骨髓抑制恢复为CHOP方案1周期,减量的BEACOP方案1周期后复发,又予Hyper-CVAD 2周期后再次达CR,予MTX及6-MP维持治疗3月,末次随访为发病后44月,为CR状态。1例患者初始治疗为BACOP方案,就诊时为ⅣB期,伴有肝功能异常,给予治疗后效果不明显,出现噬血细胞综合征后死亡。1例患者初始治疗为BEACOP方案,4疗程后达到CR,后予CHOPE方案行外周血干细胞动员并后续进行了自体干细胞移植,MTX及6-MP维持治疗3月,至今仍为CR,PFS已达96月。由此得出对于发病即为晚期的患者,单纯的CHOP化疗达到缓解的深度可能不够,增加药物依托泊苷之后的化疗方案治疗效果好,但是同时骨髓抑制也较为严重,所以建议对于早期患者可应用CHOP方案治疗,晚期患者建议使用含有依托泊苷的化疗方案或者Hyper-CVAD等强化治疗方案,而对于伴发噬血细胞综合征的患者预后较差,化疗承受力也差,目前仍无好的办法去改善预后,这与国际相关病例报道相一致[8-12]。
综上所述,SPTCL发病率低,起病主要表现为暗红色、无弹性的皮下结节,目前仍无标准治疗方案,常用的化疗方案为CHOP方案,但对晚期患者疗效不肯定;在临床实践中应用含有依托泊苷的较高强度的化疗收到较好的疗效;伴有发热及肝功能异常的患者应谨防噬血细胞综合征的发生,一旦发生治疗效果差,可试用高剂量化疗联合干细胞移植;更大样本量的多中心联合研究有待开展。
| [1] | Guenova E, Schanz S, Hoetzenecker W, et al. Systemic corticosteroids for subcutaneous panniculitis-like T-cell lymphoma[J]. Br J Dermatol, 2014, 171(4): 891–4. DOI:10.1111/bjd.13053 |
| [2] | Brown NA, Ross CW, Gudjonsson JE, et al. Subcutaneous panniculitislike T-cell lymphoma with bone marrow involvement[J]. Am J Clin Pathol, 2015, 143(2): 265–73. DOI:10.1309/AJCPVZYB19NEDXXZ |
| [3] | Swerdlow SH, Campo E, Pileri SA, et al. The 2016 revision of the World Health Organization classification of lymphoid neoplasms[J]. Blood, 2016, 127(20): 2375–90. DOI:10.1182/blood-2016-01-643569 |
| [4] | Juweid ME, Stroobants S, Hoekstra OS, et al. Use of positron emission tomography for response assessment of lymphoma: consensus of Imaging Subcommittee of International Harmonization Project in lymphoma[J]. J Clin Oncol, 2007, 25(5): 571–8. DOI:10.1200/JCO.2006.08.2305 |
| [5] | Yi L, Qun S, Wenjie Z, et al. The presenting manifestations of subcutaneous panniculitis-like T-cell lymphoma and T-cell lymphoma and cutaneous γδ T-cell lymphoma may mimic those of rheumatic diseases: a report of 11 cases[J]. Clin Rheumatol, 2013, 32(8): 1169–75. DOI:10.1007/s10067-013-2258-7 |
| [6] | Gonzalez CL, Medeiros LJ, Braziel RM, et al. T-cell lymphoma involving subcutaneous tissue. A clinicopathologic entity commonly associated with hemophagocytic syndrome[J]. Am J Surg Pathol, 1991, 15(1): 17–27. DOI:10.1097/00000478-199101000-00002 |
| [7] | Willemze R, Jaffe ES, Burg G, et al. WHO-EORTC classification for cutaneous lymphomas[J]. Blood, 2005, 105(10): 3768–85. DOI:10.1182/blood-2004-09-3502 |
| [8] | Willemze R, Jansen PM, Cerroni L, et al. Subcutaneous panniculitis-like T-cell lymphoma: definition, classification, and prognostic factors: an EORTC Cutaneous Lymphoma Group Study of 83 cases[J]. Blood, 2008, 111(2): 838–45. DOI:10.1182/blood-2007-04-087288 |
| [9] | Iqbal N, Raina V. Successful treatment of disseminated subcutaneous panniculitis-like T-cell lymphoma with single agent oral cyclosporine as a first line therapy[J]. Case Rep Dermatol Med, 2014, 2014: 201836. |
| [10] | Lee WS, Hwang JH, Kim MJ, et al. Cyclosporine A as a primary treatment for panniculitis-like T Cell lymphoma: a case with a long-term remission[J]. Cancer Res Treat, 2014, 46(3): 312–6. DOI:10.4143/crt.2014.46.3.312 |
| [11] | Chen CC, Teng CL, Yeh SP. Relapsed and refractory subcutaneous panniculitis-like T-cell lymphoma with excellent response to cyclosporine: a case report and literature review[J]. Ann Hematol, 2016, 95(5): 837–40. DOI:10.1007/s00277-016-2615-0 |
| [12] | Sakurai E, Satoh T, Akiko YA, et al. Subcutaneous panniculitis-like T-cell lymphoma(SPTCL) with hemophagocytosis(HPS): successful treatment using high-dose chemotherapy (BFM-NHL & ALL-90) and autologous peripheral blood stem cell transplantation[J]. J Clin Exp Hematop, 2013, 53(2): 135–40. DOI:10.3960/jslrt.53.135 |