 2018, Vol. 45
2018, Vol. 45文章信息
- 肾细胞癌的分子遗传学改变研究进展
- Research Progress on Molecular Genetic Alterations of Renal Cell Carcinoma
- 肿瘤防治研究, 2018, 45(4): 258-262
- Cancer Research on Prevention and Treatment, 2018, 45(4): 258-262
- http://www.zlfzyj.com/CN/10.3971/j.issn.1000-8578.2018.17.1111
- 收稿日期: 2017-09-04
- 修回日期: 2017-11-06
引用本文 |



近年来肾癌已经成为泌尿系统的高发恶性肿瘤,其发病率分别位居男性和女性常见恶性肿瘤的第7位和第10位[1]。肾癌不只是一种单一的疾病,它包括多种不同类型的肾脏肿瘤,其中肾细胞癌(RCC)约占成人肾脏恶性肿瘤的90%以上,是最常见的一种肾脏恶性肿瘤。WHO根据RCC不同的形态学和组织学特征、基因组特点将其分为不同的亚型:包括透明细胞性肾细胞癌、乳头状肾细胞癌、嫌色性肾细胞癌、集合管癌、MiT家族易位肾细胞癌、黏液样小管状和梭形细胞癌、未分类肾细胞癌等[2]。本文将重点总结几种常见RCC的基因突变、CNV和基因融合等分子遗传学改变,旨在为将来RCC的分子病理诊断奠定理论基础,也为我们进一步研究RCC的病因提供新的思路。
1 透明细胞性肾细胞癌透明细胞性肾细胞癌(ccRCC)是RCC最常见的一种病理分型,占成人肾脏恶性肿瘤的70%以上[3]。ccRCC的家族性和散发性病例都与定位于3p25的抑癌基因VHL突变密切相关,且通常发生在肿瘤发展早期[4]。Von Hippel Lindau(VHL)病就是一种由VHL基因胚系突变引起的常染色体显性遗传癌症综合征,与人体多个器官肿瘤的发展过程相关,在肾脏主要表现为ccRCC。而VHL的体细胞突变主要存在于散发性ccRCC中。据美国的肿瘤基因图谱(The Cancer Genome Atlas, TCGA)显示:在499位散发性ccRCC患者的肿瘤组织中,VHL是体细胞突变频率最高的基因(51.42%)[5]。近年来,有关VHL基因和ccRCC的相关动物模型研究也不断出现。Bailey等通过小鼠实验发现:癌基因Myc激活合并Vhl和Cdkn2a缺失的小鼠产生了类似于人类ccRCC的肾脏肿瘤[6]。Harlander等通过敲除小鼠肾小管上皮细胞的Vhl、Trp53和Rb1三个基因成功建立了ccRCC小鼠模型[7]。VHL显然已是ccRCC最主要的肿瘤相关基因,与ccRCC的发生发展都有着密不可分的联系,同时也蕴含着巨大的研究价值。随着以后更多的研究者投入到VHL与ccRCC的研究中,临床医生对于ccRCC的诊断和治疗可能会出现新的景象。除了VHL,有关ccRCC的其他肿瘤相关基因的研究也非常多。
Varela及其研究团队通过外显子测序发现:41% ccRCC的病例(92/227)中存在PBRM1的突变,并且提出了PBRM1可能是ccRCC的第二主要肿瘤相关基因[8]。然而根据TCGA数据显示:499位ccRCC病例中仅有36.08%存在PBRM1突变。相较之下TCGA的研究样本量更大,其数据似乎更具有说服力。但也可能因为存在受试者人种和地域的不同而导致结果的差异。VHL和PBRM1作为ccRCC最主要的两个肿瘤相关基因,自然也成为该研究领域的热点。Nargund等[9]通过动物模型实验发现:Vhl和Pbrm1同时缺失的小鼠肾脏容易发展成双侧、多灶性、易转移性ccRCC。另外,编码组蛋白去泛素化酶的BAP1和编码组蛋白甲基转移酶的SETD2也都是ccRCC中常见的突变基因,与VHL和PBRM1一起被列为TCGA数据库中ccRCC最常见的四个突变基因[5]。早期就有文献报道BAP1和SETD2的突变与肿瘤低生存率相关,BAP1的缺失与高级别ccRCC有关[10-11]。后来Li等[12]通过敲除人类肾小管上皮细胞的SETD2基因,结果发现SETD2基因缺失的肾小管上皮细胞没有出现细胞衰老,而是继续增殖并向ccRCC恶性转化。Miura等[13]发现在ccRCC转移位点BAP1表达缺失的患者其预后更差。Joseph等通过研究发现PBRM1和BAP1在ccRCC中存在同时表达缺失,基于PBRM1和BAP1的联合表达情况Richard将ccRCC分为4个亚型:PBRM1+BAP1+、PBRM1-BAP1+、PBRM1+BAP1-、PBRM1-BAP1-。其中PBRM1+BAP1+型的ccRCC患者预后最好,PBRM1-BAP1-预后最差[14]。这些研究结果对于预测ccRCC患者的预后情况和制定随访计划都具有重要的指导意义,当然也需要结合当地ccRCC的分子遗传学研究数据来综合评估。
此外,TCGA研究通过全外显子组测序(whole exome sequencing, WES)一共确定了19个显著突变基因(significantly mutated genes, SMGs),其中就包括上述ccRCC最常见的4个突变基因。最具有代表性的8个突变基因为:VHL、PBRM1、SETD2、KDM5C、PTEN、BAP1、MTOR和TP53,另外11个SMGs则意义较小[5]。然而ccRCC的分子遗传学改变不只是基因的突变,也包括基因组大片段拷贝数的缺失或增加。TCGA研究组运用Affymetrix SNP6.0芯片对ccRCC进行拷贝数变异(CNV)分析发现:91%的样本中存在3p的缺失,其中也包括与3p关联的四个最常见突变基因(VHL、PBRM1、SETD2和BAP1);同时还存在5q的增加(67%)和14q的缺失(45%)[5]。明显可以看出,TCGA数据库中CNV的频率比基因突变的频率更高,这对当地ccRCC病例的分子遗传学研究具有重大的指导意义。然而目前大多的研究都仅聚焦于基因突变。如果把CNV作为ccRCC的分子学标记是否会更具有特异性?未来还需更多的研究来阐明ccRCC与其CNV之间的关系。
2 乳头状肾细胞癌乳头状肾细胞癌(PRCC)是RCC的第二常见病理分型,约占成人RCC的15%[3],主要分为1型和2型两种亚型。有关PRCC的细胞遗传学分析表明2型PRCC是由1型PRCC演化而来,两种亚型具有共同的染色体变异,包括3/3q、7、12、16、17和20号染色体的三倍体表现[15]。据TCGA的CNV数据显示:在大多数1型PRCC和低级别肿瘤中,7和17号染色体的增加最常见,2、3、12、16和20号染色体的增加则相对不常见;而在大多数2型PRCC中,多条染色体的非整倍体缺失则更常见。这与之前的CNV研究结果相比更为详细和准确,不难看出PRCC中发生拷贝数增加的染色体相对比较固定,在以后的研究中或许可以考虑筛选CNV作为RCC鉴别诊断的分子标志[16]。此外,基因的胚系突变和体细胞突变也是PRCC重要的分子遗传学改变。
遗传性乳头状肾癌(HPRCC)是一种主要由MET基因发生胚系突变所引起的常染体显性遗传病,主要表现为1型PRCC。而MET的体细胞突变更常见于非遗传性PRCC中[17]。美国的TCGA研究通过WES分析确定了四个与1型PRCC相关的SMGs:包括MET、KDM6A、SMARCB1和NFE2L2。其中在81%的1型PRCC中发现有MET的体细胞突变或7号染色体的拷贝数增加[16]。这对于PRCC的临床病理分型诊断具有重要的参考价值。有文献报道,PRCC中7号染色体的增加可以导致MET拷贝数的增加,继而会引起这些肿瘤细胞中MET蛋白的过度表达,MET可能是散发性PRCC的治疗靶点[18]。最新研究表明,18%含有MET突变的晚期PRCC患者对选择性MET酪氨酸激酶抑制剂沃利替尼(savolitinib)的治疗有局部反应,而不含MET突变的PRCC患者对沃利替尼的治疗没有反应[19]。对于晚期的PRCC患者而言,这个靶向治疗研究的阶段性进展可能会给他们带来新的希望。随着研究的不断深入,晚期PRCC患者将会有更多的治疗选择。
遗传性平滑肌瘤病和肾细胞癌(HLRCC)是一种主要由编码延胡索酸水合酶的FH基因胚系突变所引起的常染体显性遗传病[20],在肾脏的主要表现为恶性程度更高的2型PRCC[21]。近年来有关2型PRCC的体细胞突变基因也不断被发现。TCGA研究通过WES分析确定了8个与2型PRCC相关的SMGs:FAT1、SETD2、NF2、KDM6A、BAP1、PBRM1、NFE2L2和TP53。TCGA的数据还显示25%的2型PRCC中存在CDKN2A改变,包括由9p21的缺失导致的CDKN2A缺失、CDKN2A启动子的高度甲基化以及CDKN2A的其他突变形式。单变量研究表明含有突变基因CDKN2A的患者其生存率比不含该突变基因的生存率低[16]。另外,PRCC的分子遗传学改变中还存在基因融合的现象。
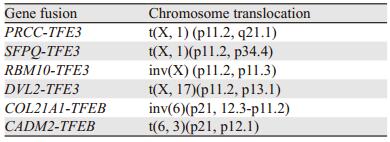
TCGA研究运用mRNA测序分析发现在PRCC中存在TFE3或TFEB的基因融合现象。其中TFE3分别与两个已知基因融合搭档(PRCC和SFPQ)和两个新的基因融合搭档(RBM10和DVL2)发生了基因融合现象,TFEB则与两个新的基因融合搭档(COL21A1和CADM2)发生基因融合[16],见表 1。新基因融合搭档的发现将可能提高临床医生对RCC基因融合现象的诊断率,继而有利于更加准确的分析和评估患者的预后情况。随着越来越多研究者的不断深入挖掘,更多未知的基因融合搭档将可能被发现。
嫌色性肾细胞癌(ChRCC)是RCC的一种相对不常见的病理分型,约占所有RCC的5%[3]。ChRCC是典型的亚二倍体表型,主要包括1、2、6、10、13、17和21号染色体[22]。后来TCGA研究运用Affymetrix SNP6.0芯片对ChRCC进行拷贝数分析发现,大多数样本(86%)中存在1、2、6、10、13和17号染色体的整条染色体单拷贝缺失,另外也观察到有3、5、8、9、11、18和21号染色体的缺失(12%~58%)[23]。不难看出TCGA的CNV数据相较于之前的研究覆盖面更广。与ccRCC和PRCC不同,ChRCC的分子遗传学改变主要表现为染色体拷贝数的改变,而发生突变的基因则相对较少。
Birt-Hogg-Dubé(BHD)综合征是一种主要由BHD胚系突变(或称为FLCN胚系突变)引发的与肾脏肿瘤发展相关的常染体显性遗传疾病,与之相关的肾脏肿瘤主要包括ChRCC和嗜酸细胞瘤[24]。然而,在大多数散发性ChRCC中没有发现BHD基因的体细胞突变,反而表现出与BHD相关肿瘤不同的基因表达谱和细胞遗传特点[25]。TCGA[23]研究通过WES对大量ChRCC样本分析发现,TP53是体细胞突变率最高的基因(32%)。TP53伴随着17号染色体的单拷贝缺失而出现杂合性缺失,从而导致该抑癌基因功能的缺失。PTEN是体细胞突变率第二的基因(9%),PTEN伴随着10号染色体的单拷贝缺失同样也出现杂合性缺失。此外,TCGA研究还发现了一些体细胞突变频率较低的基因,比如MTOR、NRAS、CDKN1A、RB1、ATM和TSC2等。Casuscelli等[26]通过38例转移性ChRCC的研究再次证实了TP53突变(58%)和PTEN突变(24%),并且发现原发肿瘤中含有TP53突变、PTEN突变和非整倍染色体复制的患者其生存率更低。Sun等[27]通过分析TCGA中含有TP53突变的ChRCC样本数据发现:HNF1B的表达均受到抑制,同时伴有HNF1B缺失和TP53突变的ChRCC患者的预后更差。这些研究都表明了ChRCC的某些分子遗传学改变与患者生存率有着密切关系,这对临床医生评估ChRCC患者的预后情况和制定随访计划都有非常重要的价值。Yang等[28]通过对源自ChRCC肉瘤样部分的新细胞系(UOK276)进行研究,不仅证实了该细胞系存在TP53的错义突变,而且发现该细胞对靶向再活化突变基因TP53的小分子治疗剂有反应。这为以后能够更加深入地研究侵袭性肉瘤样ChRCC提供了非常有价值的信息。除了ccRCC、PRCC和ChRCC三种最常见的RCC,有关一些不常见的RCC亚型的分子遗传学研究也非常多。
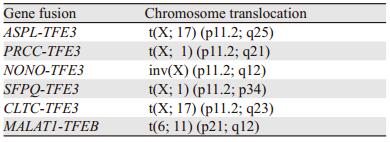
4 其他RCC亚型MiT家族易位RCC是一种不常见RCC病理分型,其分子遗传学改变主要是TFE3或TFEB的频发基因融合[29]。Xp11.2易位RCC主要涉及TFE3的基因融合,而t(6; 11)RCC主要涉及TFEB基因融合,两种类型的RCC因其在临床特征、组织形态、免疫组织化学和基因组等多方面的相似性,2013年国际泌尿病理协会将这两种RCC统称为MiT家族易位RCC[30]。2016年WHO的肾脏肿瘤分类中也沿用“MiT家族易位RCC”这个命名[2]。有研究通过反转录-聚合酶链式反应(reverse transcription-polymerase chain reaction, RT-PCR)和(或)荧光原位杂交技术(fluorescence in situ hybridization, FISH)发现,Xp11.2易位RCC最常见的两种分子遗传学改变是t(X; 1)(p11.2; q21)导致的PRCC和TFE3的基因融合[31]和t(X; 17)(p11.2; q25)导致的ASPL和TFE3的基因融合[32]。另外有文献还报道了一些比较少见的Xp11.2易位RCC的易位类型,包括:inv(X)(p11.2; q12)导致的NONO与TFE3的基因融合、t(X; 1)(p11.2; p34)导致的SFPQ与TFE3的基因融合、t(X; 17)(p11.2; q23)导致的CLTC和TFE3的基因融合。t(6; 11)RCC是一种比Xp11.2易位RCC更少见的MiT家族易位RCC亚型,迄今为止全球大约只有50个t(6; 11)RCC的病例报道,其特异性分子遗传学改变是t(6; 11)(p21; q12)导致MALAT1和TFEB基因融合[30],见表 2。目前临床病理诊断已经开始尝试将FISH检测RCC融合基因逐渐纳入适合受检人群的常规检查,随着研究者们对RCC分子水平研究的深入,临床医生对RCC的诊断、治疗和预后评估工作都将可能步入分子时代。
|
另外,还有一些罕见的RCC病理分型因其发病率极低,所以相关分子遗传学研究也比较少。黏液样小管状和梭形细胞癌(mucinous tubular and spindle cell carcinoma, MTSCC)就是一类非常少见的RCC病理分型,在2004年的WHO肾脏肿瘤分类中才被认可。通过比较基因组杂交(comparative genomic hybridization, CGH)和FISH的方法,现已证实MTSCC存在多条染色体的改变,其中包括1、4q、6、8p、9p、11q、13、14、15、18、22和X染色体的丢失,以及2、3、4、5、7、9、10、12、15、16、17、18、19、20、22和Y染色体的增加[3, 33]。集合管癌(collecting duct carcinoma, CDCA)是远侧肾单位的一种罕见肿瘤,所占比例不足全部RCC的1%。由于CDCA非常少见,有关CDCA的分子遗传学改变研究也很少[3]。Becker等[34]通过对29例CDCA病例进行CGH研究分析,发现了多条染色体的异常,包括1p、8p、9p、16p的缺失和13q的增加。
未分类肾细胞癌(unclassified renal cell carcinoma, URCC)是一种病理组织特征不属于上述任何类型的肾细胞癌,约占所有RCC的4%~5%。纪念斯隆-凯特琳癌症中心(Memorial Sloan Kettering Cancer Center, MSKCC)的研究人员对62个URCC患者样本进行体细胞突变基因检测,确定了29个经常发生体细胞突变的基因,其中突变率最高的三个基因分别为:NF(18%)、SETD(18%)和BAP1(13%)。NF在URCC中的突变率远远高于其在ccRCC、PRCC和ChRCC的突变率;而在ccRCC中突变率达75%以上的体细胞突变基因VHL,MSKCC研究人员仅在一例URCC样本中检测到有一次突变。此外,突变率在5%~10%的基因有13个:其中KMT2C、KMT2D、ATRX、DNMT3A和SMARCB1与表观遗传调节有关;MTOR、TSC1、TSC2和PTEN与mTORC1通路调节有关;KLF6、NOTCH2和TP53与转录调节有关[35]。
5 结语目前针对临床上常见RCC的分子遗传学改变的研究已经有了阶段性进展,主要表现为基因组大片段CNV、基因突变和基因融合。虽然现阶段RCC的分子诊断和治疗尚处于起步阶段,但这些研究结果都为将来RCC的分子病理诊断和靶向治疗奠定了坚实的基础。现在RCC的分子病理诊断主要局限于部分有条件的实验室,主要通过多种分子生物技术对术后肿瘤组织细胞的核内核酸进行分析,且目前数据库记录的基因突变以外显子为主。虽然现在已经有了一些关于RCC的mtDNA基因突变的初步研究,但核外核酸和内含子在不同类型的RCC中是否都有改变?它们的改变与RCC的发生发展和分型之间是否有关系?这些问题都有待进一步的实验研究来解决。另外,最近出现的肿瘤液体活检技术让RCC的术前分子病理诊断成为一种可能,将极大地推进RCC分子病理诊断的临床常规化进程。
| [1] | Siegel RL, Miller KD, Jemal A, et al. Cancer statistics, 2016[J]. CA: Cancer J Clin, 2016, 66(1): 7–30. DOI:10.3322/caac.21332 |
| [2] | Moch H, Cubilla AL, Humphrey PA, et al. The 2016 WHO Classification of Tumours of the Urinary System and Male Genital Organs-Part A: Renal, Penile, and Testicular Tumours[J]. Eur Urol, 2016, 70(1): 93–105. DOI:10.1016/j.eururo.2016.02.029 |
| [3] | Hirsch MS, Signoretti S, Cin PD, et al. Adult renal cell carcinoma: A Review of Established Entities from Morphology to Molecular Genetics[J]. Surg Pathol Clin, 2015, 8(4): 587–621. DOI:10.1016/j.path.2015.09.003 |
| [4] | Gerlinger M, Horswell S, Larkin J, et al. Genomic architecture and evolution of clear cell renal cell carcinomas defined by multiregion sequencing[J]. Nat Genet, 2014, 46(3): 225–33. DOI:10.1038/ng.2891 |
| [5] | Cancer Genome Atlas Research Network. Comprehensive molecular characterization of clear cell renal cell carcinoma[J]. Nature, 2013, 499(7456): 43–9. DOI:10.1038/nature12222 |
| [6] | Bailey ST, Smith AM, Kardos J, et al. MYC activation cooperates with Vhl and Ink4a/Arf loss to induce clear cell renal cell carcinoma[J]. Nat Commun, 2017, 8: 15770. DOI:10.1038/ncomms15770 |
| [7] | Harlander S, Schönenberger D, Toussaint NC, et al. Combined mutation in Vhl, Trp53 and Rb1 causes clear cell renal cell carcinoma in mice[J]. Nat Med, 2017, 23(7): 869–77. DOI:10.1038/nm.4343 |
| [8] | Varela I, Tarpey P, Raine K, et al. Exome sequencing identifies frequent mutation of the SWI/SNF complex gene PBRM1 in renal carcinoma[J]. Nature, 2011, 469(7331): 539–42. DOI:10.1038/nature09639 |
| [9] | Nargund AM, Pham CG, Dong Y, et al. The SWI/SNF protein PBRM1 restrains VHL-Loss-Driven clear cell renal cell carcinoma[J]. Cell Rep, 2017, 18(12): 2893–906. DOI:10.1016/j.celrep.2017.02.074 |
| [10] | Hakimi AA, Ostrovnaya I, Reva B, et al. Adverse outcomes in clear cell renal cell carcinoma with mutations of 3p21 epigenetic regulators BAP1 and SETD2: a report by MSKCC and the KIRC TCGA research network[J]. Clin Cancer Res, 2013, 19(12): 3259–67. DOI:10.1158/1078-0432.CCR-12-3886 |
| [11] | Peña-Llopis S, Vega-Rubín-de-Celis S, Liao A, et al. BAP1 loss defines a new class of renal cell carcinoma[J]. Nat Genet, 2012, 44(7): 751–9. DOI:10.1038/ng.2323 |
| [12] | Li J, Kluiver J, Osinga J, et al. Functional studies on primary tubular epithelial cells indicate a tumor suppressor role of SETD2 in clear cell renal cell carcinoma[J]. Neoplasia, 2016, 18(6): 339–46. DOI:10.1016/j.neo.2016.04.005 |
| [13] | Miura Y, Inoshita N, Ikeda M, et al. Loss of BAP1 protein expression in the first metastatic site predicts prognosis in patients with clear cell renal cell carcinoma[J]. Urol Oncol, 2017, 35(6): 386–91. DOI:10.1016/j.urolonc.2017.02.003 |
| [14] | Joseph RW, Kapur P, Serie DJ, et al. Clear cell renal cell carcinoma subtypes identified by BAP1 and PBRM1 expression[J]. J Urol, 2016, 195(1): 180–7. DOI:10.1016/j.juro.2015.07.113 |
| [15] | Gunawan B, von Heydebreck A, Fritsch T, et al. Cytogenetic and morphologic typing of 58 papillary renal cell carcinomas: evidence for a cytogenetic evolution of type 2 from type 1 tumors[J]. Cancer Res, 2003, 63(19): 6200–5. |
| [16] | Cancer Genome Atlas Research Network, Linehan WM, Spellman PT, et al. Comprehensive Molecular Characterization of Papillary Renal-Cell Carcinoma[J]. N Engl J Med, 2016, 374(2): 135–45. DOI:10.1056/NEJMoa1505917 |
| [17] | Durinck S, Stawiski EW, Pavía-Jiménez A, et al. Spectrum of diverse genomic alterations define non-clear cell renal carcinoma subtypes[J]. Nat Genet, 2015, 47(1): 13–21. DOI:10.1038/ng.3146 |
| [18] | Yin X, Zhang T, Su X, et al. Relationships between Chromosome 7 Gain, MET Gene Copy Number Increase and MET Protein Overexpression in Chinese Papillary Renal Cell Carcinoma Patients[J]. PLoS One, 2015, 10(12): e0143468. DOI:10.1371/journal.pone.0143468 |
| [19] | Gilbert JA. Savolitinib for MET-driven papillary renal cell carcinoma[J]. Lancet Oncol, 2017, 18(8): e440. DOI:10.1016/S1470-2045(17)30508-9 |
| [20] | Mehra R, Smith SC, Divatia M, et al. Emerging entities in renal neoplasia[J]. Surg Pathol, 2015, 8(4): 623–56. DOI:10.1016/j.path.2015.08.004 |
| [21] | Grubb RL 3rd, Franks ME, Toro J, et al. Hereditary leiomyomatosis and renal cell cancer: a syndrome associated with an aggressive form of inherited renal cancer[J]. J Urol, 2007, 177(6): 2074-9; discussion 2079-80. http://www.sciencedirect.com/science/article/pii/S0022534707002935 |
| [22] | Yusenko MV, Kuiper RP, Boethe T, et al. High-resolution DNA copy number and gene expression analyses distinguish chromophobe renal cell carcinomas and renal oncocytomas[J]. BMC Cancer, 2009, 9: 152. DOI:10.1186/1471-2407-9-152 |
| [23] | Davis CF, Ricketts CJ, Wang M, et al. The Somatic Genomic Landscape of Chromophobe Renal Cell Carcinoma[J]. Cancer Cell, 2014, 26(3): 319–30. DOI:10.1016/j.ccr.2014.07.014 |
| [24] | Schmidt LS, Linehan WM. Molecular genetics and clinical features of Birt-Hogg-Dubé syndrome[J]. Nat Rev Urol, 2015, 12(10): 558–69. DOI:10.1038/nrurol.2015.206 |
| [25] | Klomp JA, Petillo D, Niemi NM, et al. Birt-Hogg-Dubé renal tumors are genetically distinct from other renal neoplasias and are associated with up-regulation of mitochondrial gene expression[J]. BMC Med Genomics, 2010, 3: 59. DOI:10.1186/1755-8794-3-59 |
| [26] | Casuscelli J, Weinhold N, Gundem G, et al. Genomic landscape and evolution of metastatic chromophobe renal cell carcinoma[J]. JCI Insight, 2017, 2(12): pii:92688. DOI:10.1172/jci.insight.92688 |
| [27] | Sun M, Tong P, Kong W, et al. HNF1B loss exacerbates the development of chromophobe renal cell carcinomas[J]. Cancer Res, 2017, 77(19): 5313–26. DOI:10.1158/0008-5472.CAN-17-0986 |
| [28] | Yang Y, Vocke CD, Ricketts CJ, et al. Genomic and metabolic characterization of a chromophobe renal cell carcinoma cell line model (UOK276)[J]. Genes Chromosomes Cancer, 2017, 56(10): 719–29. DOI:10.1002/gcc.v56.10 |
| [29] | Magers MJ, Udager AM, Mehra R. MiT family translocation-associated renal cell carcinoma: a contemporary update with emphasis on morphologic, immunophenotypic, and molecular mimics[J]. Arch Pathol Lab Med, 2015, 139(10): 1224–33. DOI:10.5858/arpa.2015-0196-RA |
| [30] | Argani P. MiT family translocation renal cell carcinoma[J]. Semin Diagn Pathol, 2015, 32(2): 103–13. DOI:10.1053/j.semdp.2015.02.003 |
| [31] | Argani P, Antonescu CR, Couturier J, et al. PRCC-TFE3 renal carcinomas: morphologic, immunohistochemical, ultrastructural, and molecular analysis of an entity associated with the t(X; 1)(p11.2;q21)[J]. Am J Surg Pathol, 2012, 26(12): 1553–66. |
| [32] | Argani P, Antonescu CR, Illei PB, et al. Primary renal neoplasms with the ASPL-TFE3 gene fusion of alveolar soft part sarcoma: a distinctive tumor entity previously included among renal cell carcinomas of children and adolescents[J]. Am J Pathol, 2001, 159(1): 179–92. DOI:10.1016/S0002-9440(10)61684-7 |
| [33] | Zhao M, He XL, Teng XD. Mucinous tubular and spindle cell renal cell carcinoma: a review of clinicopathologic aspects[J]. Diagn Pathol, 2015, 10: 168. DOI:10.1186/s13000-015-0402-1 |
| [34] | Becker F, Junker K, Parr M, et al. Collecting duct carcinomas represent a unique tumor entity based on genetic alterations[J]. PLoS One, 2013, 8(10): e78137. DOI:10.1371/journal.pone.0078137 |
| [35] | Chen YB, Xu, Skanderup AJ, et al. Molecular analysis of aggressive renal cell carcinoma with unclassified histology reveals distinct subsets[J]. Nat Commun, 2016, 7: 13131. DOI:10.1038/ncomms13131 |