 2018, Vol. 45
2018, Vol. 45文章信息
- “去重”在肺癌NGS数据分析中的重要作用
- Important Role of Deduplication Method in Next Generation Sequencing Data Analysis of Non-small Cell Lung Cancer
- 肿瘤防治研究, 2018, 45(1): 1-4
- Cancer Research on Prevention and Treatment, 2018, 45(1): 1-4
- http://www.zlfzyj.com/CN/10.3971/j.issn.1000-8578.2018.17.0954
- 收稿日期: 2017-08-07
- 修回日期: 2017-11-16
引用本文 |



自2015年“精准医学”计划提出至今,高通量测序技术(next generation sequencing, NGS或二代测序)发展快速,其应用已进展至临床检测,其检测疾病的遗传学特征已成为当前精准医学的重要组成部分[1-3]。NGS流程主要包括三个部分:样本制备、测序和数据分析,其中数据分析部分又包括质控分析、序列比对、变异鉴定和变异注释。质控分析是数据分析的最重要一环,包括质量评估、去接头序列、去低质量序列、去除重复序列[4]。
目前各种NGS数据分析流程因建库方法、效率不一致而不一样,最大的差别就是有无“去重”(去除PCR扩增带来的重复序列)。多重PCR建库方法测序数据分析没有“去重”步骤,而探针杂交建库方法测序数据分析则会加入“去重”这一步骤。本研究通过比较“不去重”和“去重”两种方法分析后各NGS相关指标间的差异来进一步研究“去重”在靶向捕获NGS数据分析中的重要作用。
1 材料与方法 1.1 材料 1.1.1 样本58例NSCLC组织标本均来自广东省人民医院肺癌研究所肿瘤标本库2014—2016年收集的标本,其中10例石蜡包埋(formalin fixed paraffin-embedded, FFPE)样本、12例穿刺小样本、4例血浆样本和32例手术大样本。所有患者均知情同意并签署知情同意书。
1.1.2 主要试剂QIAGEN QIAampDNA Mini Kit和QIAGEN QIAampBlood Mini Kit试剂盒购自德国QIAGEN公司;Ion XpressTM Plus Fragment Library Kit、Ion XpressTM Barcode Adapters 1-16 Kit、Ion PITM Template OT2 200 Kit v2、Ion PITM Sequencing 200 Kit v2和Ion PITM Chip Kit v2试剂盒购自美国Applied Biosystems公司;靶向文库探针SureSelectXT Custom library由美国Agilent公司合成,SureSelectXT Reagent kit购自美国Agilent公司;Agencourt AMPure XP beads试剂购自美国Beckman公司。
1.1.3 主要仪器ABI Ion Proton二代测序仪和ABI 9700 PCR扩增仪均为美国Applied Biosystems公司产品;QIAxcel核酸分析仪为德国QIAGEN公司产品;Eppendrof 5810R离心机则为Eppendrof公司产品。
1.2 方法 1.2.1 DNA的提取应用QIAGEN QIAampDNA Mini Kit和QIAGEN QIAampBlood Mini Kit分别提取各组织样本(FFPE样本、穿刺小样本和手术大样本)的gDNA和1~4 ml血浆样本的DNA。DNA定量由Qubit分析仪分析完成。
1.2.2 设计并合成靶向捕获探针靶向区域的杂交捕获是由SureSelectXT Custom library来执行的。这个探针文库是通过SureDesign软件根据基因组hg19/GRCh37来设计的,靶向区域都是与肺癌相关的高频突变基因外显子区域。
1.2.3 NGS文库的构建NGS文库是用血浆游离DNA和组织样本的gDNA来构建的。血浆样本,提取1~2 ml血浆的游离DNA来构建文库,不需要片段化;组织样本则用50~1 000 ng gDNA通过酶切打断成100~200 bp的片段来构建文库。NGS文库是通过Ion XpressTM Plus Fragment Library Kit和Ion XpressTM Barcode Adapters 1-16 Kit来构建的,DNA片段选择和纯化回收均是通过Agencourt AMPure XP beads来完成的。片段选择后预文库进行一次11个循环的PCR扩增后再通过SureSelectXT Custom library在65℃杂交16~24 h进行靶向区域捕获,捕获到的文库在纯化和9个循环PCR扩增后即可得到靶向区域测序文库,最后分别通过QIAxcel和Qubit来检测文库片段长度和文库浓度。
1.2.4 测序及数据分析根据文库浓度用水将其稀释成12 pmol/L,用Ion PITM Template OT2 200 Kit v2将其连接到微球上,再点样到P1芯片上进行测序。测序数据通过Suite software(Life Technologies)与人类基因组Hg 19进行比对,得到相关的测序指标数据如Mapped Reads、On Target以及Uniformity等;通过Variant Caller software(Life Technologies)来检测突变,并用Integrative Genomics Viewer(IGV)来确认突变;通过Coverage Analysis software(Life Technologies)来分析平均测序深度(Mean Depth)。
1.3 统计学方法通过t检验分析“去重”与“不去重”两种方法分析得到的平均Mapped Reads、On Target、Mean Depth和Uniformity之间差异;通过方差分析比较4种不同类型样本与测序分析各指标之间的关系,通过秩和检验分析两种方法分析的EGFR、KRAS基因变异AF值间差异。P < 0.05为差异有统计学意义。
2 结果 2.1 “去重”与“不去重”两种方法分析的NGS指标间差异分析58例样本NGS数据经“不去重”和“去重”两种方法分析得到相关指标平均Mapped Reads、On Target、Mean Depth和Uniformity,见图 1。“不去重”与“去重”各组指标间差异均有统计学意义(P=0.001, P=0.001, P=0.001, P=0.001)。

|
| *: P < 0.001 图 1 NGS数据两种方法分析后相关指标差异比较 Figure 1 Differences of NGS-related indicators between duplication and de-duplication methods |
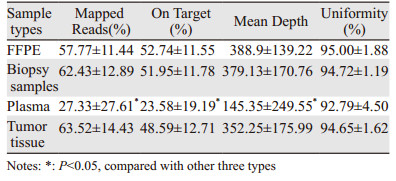
FFPE、穿刺、血浆和手术4种样本类型测序得到的NGS数据通过“不去重”方法分析时,Mapped Reads、On Target和Mean Depth 3个指标在4种类型样本间(FFPE、穿刺、血浆和手术)差异均无统计学意义,在Uniformity指标上血浆样本与其他3种类型(FFPE样本、穿刺样本、手术样本)比较差异均有统计学意义(P=0.001, P=0.001, P=0.001),见表 1。
 |
“去重”方法分析后结果正好相反,Mapped Reads指标上血浆样本与FFPE样本、穿刺样本和手术样本比较,差异均有统计学意义(P=0.006, P=0.001, P=0.001);On Target指标上血浆样本与FFPE样本、穿刺样本和手术样本比较,差异均有统计学意义(P=0.002, P=0.002, P=0.003);Mean Depth指标上血浆样本与FFPE样本、穿刺样本和手术样本组间差异均有统计学意义,但不显著(P=0.10);在Uniformity指标上则呈现出4种类型样本间差异均无统计学意义,见表 2。
 |
“去重”和“不去重”两种方法分别分析11例具有EGFR、KRAS基因突变的样本数据,去重后的AF值在数值上有增加的趋势,这种方向性变化经秩和检验分析差异有统计学意义(P=0.005)。去重是针对性消除PCR扩增所致的重复测序reads,所以去重后AF值更能反映基因突变频率的真实性,见表 3。
 |
NGS技术的不断革新加速了人们对遗传学标志物及疾病分子机制的认识。随着NGS技术发展,NGS数据也越来越庞大,数据分析也就成了制约NGS技术推广的重要瓶颈。NGS数据分析步骤繁多,包括质量评估、去接头序列、去低质量序列、去除重复序列、序列比对、变异鉴定和变异注释以及变异确认等,每一步都会对分析结果造成重大影响。其中“去除重复序列”是否会对NGS相关评价指标有影响,以及不同类型的样本之间NGS相关指标是否存在差异,尚未见相关文献报道。
本研究分析“不去重”和“去重”两种方法分析后各NGS相关指标间的差异,发现“不去重”和“去重”两种方法分析得到的各NGS相关指标间差异有统计学意义,Mapped Reads、On Target和Mean Depth 3个指标分别下降了39.85%、20.19%和52.21%,而Uniformity指标则上升了4.51%。这初步提示加入“去重”步骤对于NGS数据分析有重要作用,能够真实反映有效测序深度并改善结果的均一性。由于变异位点测序深度的变化会进一步导致等位基因频率的改变[5],故“去重”将有助于真实反映变异位点的突变频率,非常有助于临床决策。将FFPE、穿刺、血浆和手术4类样本对比分析发现,“不去重”分析时,Mapped Reads、On Target和Mean Depth 3个指标在4种类型样本间(FFPE、穿刺、血浆和手术)差异均无统计学意义,在Uniformity指标上血浆样本与其他3种类型样本(FFPE、穿刺、手术)差异均有统计学意义。而在“去重”分析时,Mapped Reads、On Target和Mean Depth 3个指标上血浆样本与FFPE样本、穿刺样本和手术样本差异均有统计学意义,在Uniformity指标上4种类型样本间差异均无统计学意义,结果正好与“不去重”分析时完全相反。两种方法分析EGFR、KRAS基因变异得到的等位基因频率有显著差异,这也进一步说明了“不去重”方法可能会掩盖掉真正的突变频率情况,从而增加临床决策难度。这些发现都提示我们只有“去重”分析才能真实的反映NGS结果。同时,两种分析方法结果都显示血浆样本明显有别于其他FFPE、穿刺和手术三类样本,这提示为获得更好的NGS分析结果,针对血浆样本,可能应建立不同于其他三类样本的建库方法以及数据分析方法。综上所述,“去重”步骤在探针杂交方法建库的NGS数据分析中具有重要的作用,能够改善结果均一性,真实反映所测样本的DNA模板数量及等位基因频率。“去重”后结果更有助于临床决策。本研究结果主要在于首先提供了“去重”对于NGS数据分析重要性的证据,后续研究将进一步论证NGS数据分析其他步骤的重要作用,为更好的完善NGS数据分析方法提供资料。
| [1] | Yang Y, Muany DM, Reid JG, et al. Clinical whole-exome sequencing for the diagnosis of mendelian disorders[J]. N Engl J Med, 2013, 369(16): 1502–11. DOI:10.1056/NEJMoa1306555 |
| [2] | Lee H, Deignan JL, Dorrani N, et al. Clinical exome sequencing for genetic identification of rare Mendelian disorders[J]. JAMA, 2014, 312(18): 1880–7. DOI:10.1001/jama.2014.14604 |
| [3] | Dewey FE, Grove ME, Pan C, et al. Clinical interpretation and implications of whole-genome sequencing[J]. JAMA, 2014, 311(10): 1035–45. DOI:10.1001/jama.2014.1717 |
| [4] | 《临床分子病理实验室二代基因测序检测专家共识》编写组. 临床分子病理实验室二代基因测序检测专家共识[J]. 中华病理学杂志, 2017, 46(3): 145–8. [ Drafting group of expert consensus on gene detection of next generation sequencing in clinical molecular pathology laboratory. Expert consensus on gene detection of next generation sequencing in clinical molecular pathology laboratory[J]. Zhonghua Bing Li Xue Za Zhi, 2017, 46(3): 145–8. ] |
| [5] | 喻东, 郭瀛军. 高通量测序临床应用中数据质量控制和分析若干问题的探讨[J]. 检验医学, 2017, 32(4): 255–61. [ Yu D, Guo YJ. Next generation sequencing in quality control and analysis of data[J]. Jian Yan Yi Xue, 2017, 32(4): 255–61. ] |