



单纯疱疹病毒(herpes simplex virus,HSV)感染是影响全球人类健康的重要问题。HSV属于疱疹病毒科α疱疹病毒亚科,是一种线性双链DNA包膜病毒,根据包膜蛋白抗原性的差别可分为HSV-1及HSV-2两种类型[1]。免疫系统正常时,HSV主要造成口腔、生殖器及眼睛等部位的感染,而免疫受损的人群还会表现出脑炎症状[2]。HSV一旦感染,就会在宿主周围神经元中建立终生可逆的潜伏期[3-4]。HSV-1会导致口腔疱疹和角膜炎爆发,同时潜伏在三叉神经节内;而HSV-2通常侵入骶神经节,引发生殖器疱疹[5]。有报告称,生殖器疱疹会使艾滋病病毒感染的危险度增加3倍左右[6-7]。目前市面上用于治疗HSV感染的药物主要是阿昔洛韦(acyclovir,ACV)及其相关的核苷类似物,但是长期使用可能会产生不良反应和病毒耐药株[3],并且目前尚无能够预防HSV的有效疫苗[6]。因此,寻找低耐药性的新型抗HSV药物尤为重要。
马兜铃酸(aristolochic acids,AAs)属于硝基菲类羧酸类化合物家族,是马兜铃、木通、细辛、广防己等植物的主要成分[8-9]。AAs具有多种药理活性,主要包括:抗病毒、抗炎、抗肿瘤等[8]。长期以来,含有AAs类化合物的中药制剂被广泛用于治疗各种疾病。但是,已有报道证明,AAs具有肾毒性,可导致马兜铃酸肾病(aristolochic acids nephropathy,AAN)。马兜铃酸I是AAs中含量最高、毒性最强的成分,小鼠以20 mg·kg-1剂量给药5 d即出现明显肾损伤现象[9]。综上,AAs用于临床时应严格把控剂量,合理使用。
目前为止,鲜有对AAs抗疱疹病毒活性的报道,本研究对AAs的体内外抗HSV作用及其作用机制进行了初步探究。
1 材料与方法 1.1 材料 1.1.1 药物AAs(313-67-7)购于陶素化工有限公司(中国上海)。ACV购于湖北午时药业股份有限公司。
1.1.2 细胞和病毒非洲绿猴肾细胞(Vero)培养于MEM培养基,添加10%胎牛血清(ExCell Bio,中国),青霉素(100 kU·L-1),链霉素(100 mg·L-1),培养于37 ℃ 5% CO2的培养箱中。HSV-1 F株购于ATCC(美国)。HSV-2 333株来源于中国科学院武汉病毒研究所。
1.1.3 试剂4%多聚甲醛(上海源叶生物科技有限公司);5%结晶紫染色液、5×蛋白上样缓冲液、PMSF、RIPA裂解液、碱性磷酸酶检测试剂盒、苏木素-伊红染色试剂盒购于碧云天生物技术有限公司,胎牛血清从ExCell Bio公司购买,琼脂粉、MEM干粉购于Gibco公司,0.25% 胰酶、双抗购于新赛美生物科技有限公司,β-actin抗体从Cell Signalling Technology公司购买,HSV gD、gB、ICP27抗体购于Abcam公司,RNA提取试剂盒购于Aid lab公司,One Step TB Green® Prime ScriptTM RT-PCR Kit Ⅱ试剂盒购于TaKaRa公司。
1.1.4 仪器Ⅱ级B2型生物安全柜购于新加坡Esco公司;CO2培养箱及台式离心机购于美国Thermo公司,酶标仪购于美国伯腾仪器有限公司;蛋白电泳和转膜设备从美国Bio-Rad公司购买;倒置相差显微镜购于日本Olympus公司;ABI 7500型定量PCR仪购于美国应用生物系统公司。
1.2 方法 1.2.1 细胞毒性实验接种Vero细胞于96孔板中,至其长满单层,吸弃原培养液,将梯度稀释的AAs(终浓度200、180、160、140、120、100、50、25 μmol·L-1)加入96孔板,每个浓度3个复孔,同时设有空白对照组。37 ℃ 5% CO2培养24 h后,吸弃药物,加入4%多聚甲醛室温固定15 min,吸弃,加入5%结晶紫室温染色15 min,清洗晾干后用酶标仪测定A540nm值。
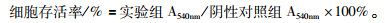
|
将AAs梯度稀释(终浓度50、25、12.5、6.25 μmol·L-1),对HSV-1/HSV-2感染的细胞进行全过程给药(预处理细胞、预处理病毒、吸附时、吸附后)。预处理细胞(Pre+Cell):AAs于37 ℃作用细胞1 h,吸弃,37 ℃吸附HSV(MOI=0.1)1 h,换成维持液;预处理病毒(Pre+Virus):AAs与HSV于37 ℃混合孵育1 h后,加入吸弃原培养基的96孔板中,37 ℃作用1 h,换成维持液;吸附时(adsorption)给药:AAs与HSV(MOI=0.1)混合后加入96孔板中,于37 ℃作用1 h,换成维持液;吸附后(post-adsorption)给药:HSV于37 ℃吸附细胞1 h,换成含有AAs的维持液。每个浓度设有3个复孔,同时设有病毒对照组和空白对照组。30 h后按“1.2.1”方法检测A540nm,计算CPE抑制率。CPE抑制率/%=(实验组A540nm-病毒对照组A540nm)/(空白对照组A540nm-病毒对照组A540nm)×100%。
1.2.3 空斑实验接种Vero细胞于12孔板中,至其长满单层。按照“1.2.2”中4种不同方式分别加入AAs(终浓度20 μmol·L-1);或者先用HSV-1(MOI=0.1)于37 ℃吸附细胞1 h,吸弃,然后加入不同浓度的AAs(终浓度40、20、10、5 μmol·L-1)处理细胞;此外,先用HSV-1(MOI=0.1)于37 ℃吸附细胞1 h,之后于吸附后不同时间段(0~2、2~4、4~6、6~8、8~10、10~12、0~6、0~12 h)加入AAs(终浓度20 μmol·L-1)。24 h后收取上清,同时设病毒对照组。收取的上清稀释10~1 000倍,于37 ℃吸附细胞1 h,吸弃,加入配制好的覆盖培养基,待其冷却凝固后,倒置于37 ℃培养箱中。至其出斑后,固定染色,进行空斑计数。
1.2.4 间接免疫荧光实验接种Vero细胞于3.5 cm玻底皿,待细胞长至密度30%~40%时,用AAs(终浓度20 μmol·L-1)按照1.2.2中4种不同作用方式处理HSV-1(MOI=1.0),置于37 ℃培养箱中培养8 h。然后用PBS清洗细胞,4%多聚甲醛室温固定15 min,PBS清洗2 min,0.25% Triton X-100通透10 min,PBS清洗2 min,2% BSA于37 ℃封闭1 h,PBS清洗3次,加入ICP5的抗体(1 ∶100稀释)于4 ℃孵育16 h,清洗后加入DyLight 649®偶联二抗于4 ℃孵育4 h,清洗3次,DAPI处理10 min,清洗后进行共聚焦成像,并使用ImageJ(NIH)(美国)进行分析。
1.2.5 膜融合抑制实验接种Vero细胞于12孔板中,待细胞长满单层,用HSV-1/HSV-2(MOI=0.1)于37 ℃感染细胞1 h,然后将含有不同浓度AAs(终浓度20、10、5、2.5 μmol·L-1)的维持液加入相应孔中,同时设有病毒对照组和空白对照组。12 h后进行苏木精-伊红染色(HE染色),显微镜下拍照后用ImageJ处理图像。
1.2.6 Western blot实验接种Vero细胞于6孔板中,待细胞长满单层,用HSV-1/2(MOI=0.1)于37 ℃感染细胞1 h,然后将含有不同浓度AAs(终浓度20、10、5、2.5 μmol·L-1)的维持液加入相应孔中,同时设有病毒对照组和空白对照组,16 h后收样;Vero细胞接种于12孔板中,HSV-1(MOI=0.1)于37 ℃吸附细胞1 h,于吸附后不同时间段(0~2、2~4、4~6、6~8、8~10、10~12、0~6、0~12 h)加入AAs(终浓度20 μmol·L-1),16 h后收样;接种Vero细胞于6孔板中,待细胞长满单层,用HSV-1(MOI=0.1)于37 ℃感染细胞1 h,然后将含有不同浓度AAs(终浓度80、40、10、5 μmol·L-1)的维持液加入相应孔中,同时设有病毒对照组和空白对照组,24 h后收样。每孔加入100 μL细胞裂解液(800 μL RIPA,200 μL 5×蛋白上样缓冲液,10 μL PMSF),冰上裂解17 min,将裂解液刮下后转移到EP管中,100 ℃煮样20 min,放在-20 ℃冰箱备用。用BCA试剂盒测定提取的蛋白浓度后进行SDS-PAGE电泳,将电泳胶通过半干法转移到NC膜上,将NC膜用5%的奶粉于4 ℃封闭过夜,1×TBST洗涤3次,用HSV-1/HSV-2 ICP27和gB蛋白抗体、抗磷酸化p38 MAPK和NF-κB的抗体稀释液(1 ∶1 000稀释)于37 ℃孵育2 h,继续洗涤3次,然后用二抗稀释液(1 ∶5 000稀释)于37 ℃孵育2 h。用碱性磷酸酶检测试剂盒显色,拍照后用ImageJ处理图像。
1.2.7 实时荧光定量PCR接种Vero细胞于6孔板中,待细胞长满单层,用HSV-1/2(MOI=0.1)于37 ℃感染细胞1 h,然后将含有不同浓度AAs(终浓度20、10、5、2.5 μmol·L-1)的维持液加入相应孔中,同时设有病毒对照组和空白对照组。16 h后用RNA提取试剂盒提取总RNA。对提取的RNA进行浓度检测后,利用Applied Biosystems 7500 Fast Real Time PCR System,按照One Step TB Green® Prime ScriptTM RT-PCR Kit Ⅱ试剂盒的说明书进行操作。引物序列:HSV-1(gD) mRNA Forward primer:5′-AGCAGGGGTTAGGGAGTTG-3′; HSV-1(gD) mRNA Reverse primer:5′-CCATCTTGAGAGAGGCATC-3′; HSV-2(gD) mRNA Forward primer:5′-AGCATCCCGATCACTGTGTACTA-3′; HSV-2(gD) mRNA Reverse primer:5′-GCGATGGTCAGGTTGTACGT-3′; Actin mRNA Forward primer:5′-CTCCATCCTGGCCTCGCTGT-3′; Actin mRNA Reverse primer:5′-GCTGTCACCTTCACCGTTCC-3′。
1.2.8 无病毒膜融合实验分别接种Vero细胞与BSRT7/5细胞于6孔板中,待细胞长至50%~70%,转染质粒。Vero细胞共转染pcDNA3.1(+) HSV-2-gB、pcDNA3.1(+) HSV-1-gD、pcDNA3.1(+) HSV-1-gH、pcDNA3.1(+) HSV-1-gL、pCAGGS-T7 luc质粒(以下简称gB、gD、gH、gL、T7 luc),转染比例为1 ∶1 ∶2 ∶2 ∶4,BSRT7/5细胞共转染gB、gD、gH、gL质粒,转染比例为1 ∶1 ∶2 ∶2。转染后24 h,将两种细胞分别消化,1 ∶1混合接种于新的孔板中,同时给药组加入不同浓度的药物(杨梅素终浓度10、5 μmol·L-1,AAs终浓度20、10、5 μmol·L-1),于37 ℃培养42 h后利用荧光素酶检测试剂盒进行检测。
1.2.9 动物实验使用3~4周龄的BALB/c雌鼠,体质量(14~15) g,动物许可证号SCXK(鲁)20190003。随机分为5组,每组12只:空白对照组,病毒对照组,阳性药ACV(10 mg·kg-1·d-1)组,AAs高剂量(10 mg·kg-1·d-1)组及AAs低剂量(5 mg·kg-1·d-1)组。采用滴鼻方式接毒[13],接毒后4 h给药,连续给药5 d。接毒后72 h解剖,取小鼠的肺及脊髓,通过空斑实验及实时荧光定量PCR验证药物在体内抗HSV-1的作用效果。以接毒日为起始日,连续14 d观察小鼠临床症状,每天记录小鼠体质量变化及生存情况。
1.2.10 统计学分析数据分析及绘制统计图表均采用GraphPad Prism 8.0统计软件。各组数据先进行正态性和方差齐性检验,采用单因素方差分析和t检验比较组间的差异。
2 结果 2.1 AAs的细胞毒性实验将AAs稀释为终浓度25、50、100、120、140、160、180、200 μmol·L-1,分别处理细胞24 h,结果如Fig 1所示,AAs在25 μmol·L-1时,细胞存活率接近80%;随着浓度逐渐增大,细胞存活率逐渐下降,当浓度达到180 μmol·L-1时细胞存活率下降至50%左右。经计算,AAs对Vero细胞的半数细胞毒性浓度CC50值为193.1 μmol·L-1。

|
| Fig 1 Cytotoxicity of aristolochic acids on Vero cells(x±s, n=3) |
我们进一步利用CPE抑制实验评价了AAs全过程给药对于HSV-1和HSV-2的抑制作用。结果如Fig 2所示,AAs在6.25~50 μmol·L-1的浓度范围内对HSV-1及HSV-2感染均有明显抑制作用。其中,AAs对HSV-1感染的抑制效果略优于HSV-2,其IC50值约为11.12±3.06 μmol·L-1,治疗指数为17.37(Tab 1)。同时设置ACV作为阳性对照组,结果显示AAs抑制HSV-1感染的IC50值略低于ACV,但抑制HSV-2感染的IC50值略高于ACV,说明AAs对HSV-1的抑制作用略优于ACV。根据CC50和IC50值计算治疗指数(SI),结果表明ACV的SI值分别是AAs的7倍和10倍,说明AAs的药物安全性相对较差,其药物毒性问题仍需进一步解决。本研究中体外实验的AAs使用剂量以终浓度40 μmol·L-1为例,在Vero细胞上给药100 μL时AAs含量约为0.014 mg,相当于体内实验中一只15 g的小鼠以肾毒性剂量(20 mg·kg-1)给药时AAs含量(0.3 mg)的1/20。

|
| Fig 2 Inhibition of HSV-1/HSV-2 infection by aristolochic acids on Vero cells(x±s, n=3) |
| CC50/μmol·L-1 | HSV-1 IC50/μmol·L-1 | SI(CC50/IC50) | HSV-2 IC50/μmol·L-1 | SI(CC50/IC50) | |
| Aristolochic acids | 193.1±36.6 | 11.12±3.06 | 17.37 | 25.42±4.36 | 7.60 |
| Acyclovir | 1387±278 | 11.36±1.98 | 122.10 | 17.6±2.88 | 78.80 |
AAs以4种作用方式作用于HSV感染的Vero细胞后,24 h收取上清液。用收取的上清液进行空斑实验,检测病毒滴度,进一步评价AAs对病毒增殖的抑制作用。结果如Fig 3所示,预处理细胞组、预处理病毒组、吸附时给药组对病毒滴度降低的作用不大,但是吸附后给药组明显降低病毒滴度,在20 μmol·L-1的浓度下几乎完全抑制了HSV的增殖。这说明AAs主要作用于病毒吸附后的感染阶段。

|
| Fig 3 Results of plaque assay of aristolochic acids in different treatment conditions on HSV infection(x±s, n=3) A and B: Plaque assay of HSV-1 titers in aristolochic acids treated Vero cells under different treatment conditions; C and D: Plaque assay of HSV-2 titers in aristolochic acids treated Vero cells under different treatment conditions. *P < 0.05, **P < 0.01 vs virus control. |
我们进一步利用间接免疫荧光实验评价AAs在不同作用方式下对HSV-1病毒蛋白gD表达的抑制作用。AAs按照上述4种不同作用方式处理细胞或病毒,8 h后利用免疫荧光检测病毒gD蛋白的表达情况。结果如Fig 4A所示,无药物处理的病毒组可以检测到大量gD蛋白的红色荧光,预处理细胞、预处理病毒及吸附时给药组的红色荧光相对于病毒组大幅减少,而吸附后给药组的红色荧光最弱。因此,AAs在吸附后给药对于HSV蛋白表达抑制作用最强。

|
| Fig 4 Inhibition of aristolochic acids on viral titer in HSV infected Vero cells(x±s, n=3) A: Immunofluorescence assay of HSV-1 gD protein in aristolochic acids treated Vero cells under different treatment conditions; B: Plaque assay of HSV-1 titers in aristolochic acids or acyclovir treated Vero cells.*P < 0.05, **P < 0.01 vs virus control. |
此外,我们还评价了不同浓度的AAs以吸附后给药的方式作用于Vero细胞对于HSV增殖的抑制作用,设置ACV作为阳性对照组。即在HSV感染24 h后收取上清液,用收取的上清稀释液进行空斑实验,检测病毒滴度。结果如Fig 4B所示,AAs在2.5~40 μmol·L-1的浓度范围内能明显抑制HSV在Vero细胞中的增殖,且呈现明显的剂量依赖性。同时,与ACV组进行对比,在药物浓度为10 μmol·L-1时,AAs处理可使病毒滴度下降约2.7个Lg(PFU·mL-1),而ACV处理使病毒滴度下降约2.2个Lg(PFU·mL-1),可见AAs抑制HSV-1增殖效果略优于ACV,这进一步说明AAs主要通过影响病毒吸附后的感染事件来抑制HSV的增殖。
2.5 AAs对HSV诱导的膜融合的抑制作用HSV进入Vero细胞,需要病毒-细胞膜融合,这种融合需要HSV包膜糖蛋白gB、gD、gH/L异源二聚体的参与[10-11]。Vero细胞感染HSV-1或HSV-2后,用不同浓度的AAs(5、10、20 μmol·L-1)处理,30 h后对细胞进行HE染色,结果如Fig 5A-D所示,无药物处理的病毒对照组出现明显的膜融合现象(红色箭头指示合胞体形成),而不同浓度AAs处理后,膜融合现象受到明显抑制。其中20 μmol·L-1时抑制现象最明显,这种抑制作用同样具有剂量依赖性(Fig 5B和5D)。

|
| Fig 5 Inhibition of aristolochic acids on membrane fusion induced by HSV(x±s, n=3) A and B: Aristolochic acid inhibits syncytium formation mediated by HSV-1; C and D: Aristolochic acid inhibits syncytium formation mediated by HSV-2; E and F: Virus-free fusion inhibition assay of myricetin (E) or aristolochic acids (F) treated HSV infected Vero cells.*P < 0.05, **P < 0.01 vs virus control. |
HSV的gB、gD、gH、gL蛋白对于膜融合现象的产生起着关键作用,于是我们进一步利用无病毒细胞-细胞膜融合实验(分子膜融合实验)检测AAs是否与gB、gD、gH、gL 4种蛋白直接作用而抑制膜融合现象。由于4种HSV包膜蛋白(gB、gD、gH/L异源二聚体)对于细胞-细胞融合现象的产生必要且充分[11]。于是,我们将Vero细胞共转染pcDNA3.1(+)-gB、pcDNA3.1(+)-gD、pcDNA3.1(+)-gH、pcDNA3.1(+)-gL及pCAGGS-T7 luc这5种质粒,BSRT7/5细胞共转染pcDNA3.1(+)-gB、pcDNA3.1(+)-gD、pcDNA3.1(+)-gH、pcDNA3.1(+)-gL 4种质粒后24 h,将两种转染细胞混合,同时加入不同浓度的药物,培养42 h后利用荧光素酶检测试剂盒进行定量检测。前期的研究中我们发现,杨梅素可与病毒gD蛋白质直接作用从而对膜融合现象产生抑制[12]。因此,本研究中以杨梅素作为膜融合抑制的阳性对照药。结果如Fig 5E和5F所示,杨梅素浓度为5 μmol·L-1时即可减少约70%由gB、gD、gH/L引起的膜融合现象,当其10 μmol·L-1时几乎可以完全抑制膜融合,与之前的报道是一致的。但是,用不同浓度AAs处理后,膜融合现象并未被抑制。因此,AAs并非通过与gB、gD、gH/L 4种蛋白直接相互作用对膜融合现象产生抑制,而是通过抑制HSV的其它吸附后的感染事件来间接抑制HSV诱导的膜融合。
2.6 AAs在吸附后不同作用时间段对HSV的抑制作用通过上述实验可以看出,AAs主要在吸附后阶段对HSV感染产生抑制作用,于是,我们进一步利用Western blot及空斑实验对AAs在HSV感染后不同时间阶段给药的抑制活性进行了评价。如Fig 6所示,Western blot及空斑实验结果基本一致,AAs在吸附后2~4 h及4~6 h给药可明显降低病毒滴度,在6~8、8~10、10~12 h几乎没有抑制作用。另外,在0~6 h或0~12 h用AAs处理对病毒增殖的抑制作用更为明显。因此,AAs在HSV感染后的早期阶段即2~6 h作用效果较好。

|
| Fig 6 Inhibition of aristolochic acids on HSV in different time periods after adsorption(x±s, n=3) A and B: Western blot analysis of HSV-1 ICP27 expression after treated with aristolochic acids at different time intervals. Quantification of immunoblot for the ratio of ICP27 to β-actin was also shown (B); C: Plaque assay of HSV-1 titers in Vero cells treated with aristolochic acids at different time intervals.*P < 0.05, **P < 0.01 vs virus control. |
既然AAs主要作用于病毒吸附后的2~6 h的感染阶段,我们进一步利用Western blot和RT-PCR评价了AAs对病毒蛋白表达和mRNA表达的抑制作用。首先,在Vero细胞感染HSV-1或HSV-2后,按照吸附后给药的方式,用AAs(2.5、5、10、20 μmol·L-1)处理细胞,16 h后收取蛋白样品并进行SDS-PAGE电泳。我们选择HSV-1蛋白ICP27及HSV-2蛋白gB进行检测。结果如Fig 7A-D所示,AAs在2.5~20 μmol·L-1的浓度范围内对HSV-1的ICP27蛋白和HSV-2的gB蛋白的表达都具有明显的抑制作用。总之,AAs对HSV感染后蛋白的表达具有明显抑制作用,且具有剂量依赖性。

|
| Fig 7 Inhibition of aristolochic acids on HSV protein and mRNA expression(x±s, n=3) A and B: Western blot analysis of HSV-1 ICP27 expression after treated with aristolochic acids; C and D: Western blot analysis of HSV-2 gB expression after treated with aristolochic acids; E and F: Influence of aristolochic acids on mRNA expression of HSV-1 (E) or HSV-2 (F) gD protein, respectively. *P < 0.05, **P < 0.01 vs virus control. |
此外,我们还利用实时荧光定量PCR法评价了AAs对病毒gD蛋白mRNA的表达的影响。结果如Fig 7E和7F所示,在AAs浓度为5 μmol·L-1以上时,对HSV-1和HSV-2的gD蛋白的mRNA表达具有明显的抑制作用,与Western blot实验结果基本一致,这说明AAs可以在病毒吸附后的复制过程中产生抑制作用,且这种抑制作用具有明显的剂量依赖性。
2.8 AAs对p38 MAPK/NF-κB信号通路蛋白表达的影响前期实验发现,AAs可以抑制HSV吸附后的某些步骤,降低HSV的mRNA和蛋白表达,因此我们进一步探讨AAs是否可以影响HSV感染细胞的相关信号通路。
丝裂原活化蛋白激酶(MAPK)是丝氨酸-苏氨酸激酶,介导与一系列细胞活动相关的细胞内信号,包括细胞增殖、分化、存活、死亡等[13]。p38 MAPK与细胞炎症、凋亡、生长有关,据报道,HSV-1感染细胞p38 MAPK的激活可促进病毒的复制[14]。NF-κB可以调控炎性细胞因子和趋化因子的表达,在抑制细胞凋亡,以及免疫和炎症反应的调节中发挥重要作用[15-16]。据报道,HSV感染可以激活NF-κB,从而提高病毒复制的效率[16]。在此基础上,我们检测了在HSV-1感染后,AAs对细胞p38 MAPK及NF-κB信号蛋白的影响。
如Fig 8A、B所示,在细胞感染HSV-1 24 h后,病毒组中的p38 MAPK蛋白磷酸化水平约为正常对照组2.1倍。然而,AAs(20、40、80 μmol·L-1)处理后,p-p38 MAPK水平从正常对照组的2.1倍明显下降到1.2、0.8和0.7倍,证明AAs能够明显降低由HSV感染导致的p38 MAPK的活化。

|
| Fig 8 Effect of aristolochic acids on p38 MAPK and NF- κB activation after HSV infection(x±s, n=3) A~C: Western blot analysis of the effects of aristolochic acids on p38 MAPK/NF-κB activation in HSV-1 infected Vero cells. Quantification of immunoblot for the ratio of p-p38 MAPK (B) or p-NF-κB (C) to β-actin was also shown.*P < 0.05, **P < 0.01 vs virus control; ##P < 0.01 vs mock. |
NF-κB是p38 MAPK通路的下游信号之一。如Fig 8A、8C所示,在细胞感染HSV-1 24 h后,病毒组中的NF-κB蛋白磷酸化水平约为正常对照组1.5倍。然而,AAs(40、80 μmol·L-1)处理后,NF-κB水平从正常对照组的1.5倍明显下降到1和0.7倍,证明AAs能够明显降低由HSV感染导致的NF-κB的活化,抑制HSV的复制。因此,细胞p38 MAPK/NF-κB信号通路可能参与AAs体外抗HSV作用。
2.9 AAs的体内抗HSV作用我们进一步利用HSV-1感染BALB/c小鼠建立HSV感染的小鼠肺炎和脑炎模型评价AAs的体内抗HSV作用,并与阳性药ACV进行对比分析。据报道,感染HSV的小鼠以ACV(10 mg·kg-1)处理7 d即可明显降低死亡率[17],本研究设置阳性药ACV组剂量为10 mg·kg-1。基于AAs肾毒性剂量约为20 mg·kg-1,同时为了控制实验变量,在相同剂量下比较受试药物AAs的疗效,选择1/4和1/2肾毒性剂量(5、10 mg·kg-1)的AAs进行体内研究。结果如Fig 9A所示,与空白组相比,病毒组小鼠体质量在接毒后持续下降,并始终轻于原始体质量。阳性药ACV组(10 mg·kg-1)体质量前3 d缓慢下降,d 3后开始持续回升;AAs高剂量组(10 mg·kg-1)前9 d体质量一直缓慢下降,d 9后开始缓慢回升,但仍低于空白组;AAs低剂量组(5 mg·kg-1)前7 d体质量一直缓慢下降,d 7后开始缓慢回升,但仍低于空白组。

|
| Fig 9 Inhibition effect of aristolochic acids on HSV infection in vivo A: The body weights of mice were monitored daily for 14 days and are expressed as a percentage of the initial value(x±s, n=12); B: The survival rates of mice are expressed as percentage of survival, evaluated daily for 14 days(x±s, n=12). **P < 0.01 vs virus control group (placebo); The viral titres in lungs (C) or spinal cords (D) were evaluated by plaque assay and quantitative RT-PCR assay on day 3 p.i., respectively(x±s, n=3)*P < 0.05, **P < 0.01 vs virus control. |
而Fig 9B的小鼠生存率结果表明,病毒组小鼠在接毒后d 2开始出现死亡,并且在d 6时生存率降至30%;而阳性对照药ACV组(10 mg·kg-1)在接毒后d 1开始出现死亡,d 9时生存率降至70%;AAs高剂量组(10 mg·kg-1)在接毒后未出现死亡现象;AAs低剂量组(5 mg·kg-1)在接毒后d 5开始出现死亡现象,在d 8时生存率降低至60%。AAs处理后,与病毒组相比小鼠体质量上升趋势不明显,且AAs高剂量组体质量下降程度大于低剂量组,推测AAs可能在10 mg·kg-1时就具有肾毒性。但是,AAs处理组的生存率明显提高(P < 0.01),这说明AAs处理能够明显改善小鼠感染HSV-1后的生存状况,提高小鼠生存率。
此外,我们利用空斑实验检测肺部组织的病毒载量,并利用实时荧光定量PCR检测脊髓组织中的病毒载量,以此来评价AAs对于HSV体内增殖的影响。结果如Fig 9C所示,与病毒组相比,阳性药ACV组肺病毒载量下降了0.3个lg(PFU·mL-1),AAs高剂量组肺病毒载量下降了0.9个lg(PFU·mL-1),而AAs低剂量组下降了1.3个lg(PFU·mL-1)。AAs低剂量组优于高剂量组,且都略优于阳性药组。另外,Fig 9D的结果表明,与病毒组相比,阳性药ACV降低了小鼠脊髓组织中的约80%的病毒载量,AAs高剂量组降低了约70%的病毒载量,AAs低剂量组则降低了约90%。AAs低剂量组优于高剂量组,且效果都优于阳性药ACV组。上述结果表明,AAs在体内动物水平上能够明显降低小鼠组织中的病毒载量,且效果优于阳性药ACV。
3 讨论AAs具有广泛的生物活性和药理作用,主要有抗病毒、抗肿瘤、抗菌、抗炎、镇痛以及对心血管的药理作用[7-8]。目前为止,尚无关于AAs抗HSV作用的报道。本研究前期利用Vero细胞模型发现AAs具有抗HSV-1及HSV-2的活性,且对Vero细胞毒性较低。本研究从体外和体内两个方向入手,对AAs抗HSV的活性进行了探究。
我们体外研究发现,吸附后给药能够明显抑制HSV感染导致的细胞病变效应,同时无病毒膜融合现象不能被AAs抑制,提示其可能作用于病毒进入或后续感染步骤,且作用靶点并非HSV gB、gD、gH/gL蛋白。病毒吸附后不同时间范围给药实验表明AAs在病毒吸附后的早期阶段即2~6 h作用效果较好。吸附后加入AAs能够抑制HSV感染诱导的膜融合且能够抑制ICP27、gB、gD蛋白的表达,再次证明AAs能够抑制病毒吸附后的感染过程。同时,AAs能明显减少病毒感染后p38 MAPK和NF-κB蛋白的磷酸化程度,这可能与病毒感染后AAs抑制HSV复制的过程有关。
此外,利用HSV-1感染BALB/c小鼠建立HSV感染的小鼠肺炎和脑炎模型评价AAs的体内抗HSV作用。研究发现腹腔注射AAs治疗HSV-1感染的小鼠可明显提高小鼠的存活率;但经AAs处理后,小鼠体质量无明显上升,可能与其毒性有关。肺匀浆空斑实验及脊髓匀浆实时荧光定量PCR实验看出,AAs可明显抑制HSV在肺和脊髓中的增殖,效果略优于阳性药ACV。
综上所述,AAs具有很好的体内外抗HSV感染作用,其主要作用于病毒吸附后的感染阶段,但其具体作用靶点仍有待进一步探究。AAs对HSV在小鼠肺和脊髓中的增殖有明显抑制作用,但其体内毒性问题仍待解决。本研究首次揭示了AAs的抗HSV作用,并对其抗HSV作用机制进行了初步探究。研究结果表明,AAs具有开发成抗HSV药物的潜力,但需要严格控制使用剂量,值得后续进一步研究。
| [1] |
Rechenchoski D Z, Faccin-Galhardi L C, Linhares REC, et al. Herpesvirus: An underestimated virus[J]. Folia Microbiol (Praha), 2017, 62(2): 151-6. doi:10.1007/s12223-016-0482-7 |
| [2] |
Harris K D. Herpes simplex virus keratitis[J]. Home Healthc Now, 2019, 37(5): 281-4. doi:10.1097/NHH.0000000000000791 |
| [3] |
Treml J, Gazdová M, Mejkal K, et al. Natural products-derived chemicals: Breaking barriers to novel anti-HSV drug development[J]. Viruses, 2020, 12(2): 154. doi:10.3390/v12020154 |
| [4] |
罗卓, 颜畅, 李怡芳, 等. Ⅰ型单纯疱疹病毒潜伏感染及其应激复发[J]. 中国药理学通报, 2017, 33(9): 1185-90. Luo Z, Yan C, Li Y F, et al. The latent infection of HSV-1 and stress-induced reactivation[J]. Chin Pharmacol Bull, 2017, 33(9): 1185-90. doi:10.3969/j.issn.1001-1978.2017.09.001 |
| [5] |
Porter D. What is herpes keratitis? American academy of ophthalmology[OL]. 2018[2021-12-17]. https://www.aao.org/eye-health/diseases/herpes-keratitis.
|
| [6] |
Kim H C, Lee H K. Vaccines against genital herpes: Where are we?[J]. Vaccines, 2020, 8(3): 420. doi:10.3390/vaccines8030420 |
| [7] |
Crisci E, Svanberg C, Ellegrd R, et al. HSV-2 cellular programming enables productive HIV infection in dendritic cells[J]. Front Immunol, 2019, 10: 2889. doi:10.3389/fimmu.2019.02889 |
| [8] |
Sidorenko V S. Biotransformation and toxicities of aristolochic acids[J]. Adv Exp Med Biol, 2020, 1241: 139-66. |
| [9] |
崔媛, 李海山, 宋乃宁, 等. 马兜铃酸Ⅰ影响小鼠肝脏脂肪酸β氧化和糖代谢以及TCA循环的代谢组学研究[J]. 中国药物警戒, 2019, 16(8): 18. Cui Y, Li H S, Song N N, et al. Metabonomics reveals that aristolochic acid I affects β-oxidation of fatty acids, glucose metabolism and the TCA cycle in the mice liver[J]. Chin J Pharmcovigilance, 2019, 16(8): 18. |
| [10] |
Barrow E, Nicola A V, Liu J. Multiscale perspectives of virus entry via endocytosis[J]. Virol J, 2013, 10: 177. doi:10.1186/1743-422X-10-177 |
| [11] |
Weed D J, Nicola A V. Herpes simplex virus membrane fusion[J]. Adv Anat Embryol Cell Biol, 2017, 223: 29-47. |
| [12] |
Li W, Xu C, Hao C, et al. Inhibition of herpes simplex virus by myricetin through targeting viral gD protein and cellular EGFR/PI3K/Akt pathway[J]. Antiviral Res, 2020, 177: 104714. doi:10.1016/j.antiviral.2020.104714 |
| [13] |
Kim E K, Choi E J. Pathological roles of MAPK signaling pathways in human diseases[J]. Biochim Biophys Acta, 2010, 1802(4): 396-405. doi:10.1016/j.bbadis.2009.12.009 |
| [14] |
Zachos G, Koffa M, Preston C M, et al. Herpes simplex virus type 1 blocks the apoptotic host cell defense mechanisms that target Bcl-2 and manipulates activation of p38 mitogen-activated protein kinase to improve viral replication[J]. J Virol, 2001, 75(6): 2710-28. doi:10.1128/JVI.75.6.2710-2728.2001 |
| [15] |
Song Y H, Chai Q, Wang N L, et al. X-rays induced IL-8 production in lung cancer cells via p38/MAPK and NF-κB pathway[J]. Int J Radiat Biol, 2020, 96(11): 1374-81. |
| [16] |
Gregory D, Hargett D, Holmes D, et al. Efficient replication by herpes simplex virus type 1 involves activation of the IkappaB kinase-IkappaB-p65 pathway[J]. J Virol, 2004, 78(24): 13582-90. |
| [17] |
Quenelle D C, Birkmann A, Goldner T, et al. Efficacy of pritelivir and acyclovir in the treatment of herpes simplex virus infections in a mouse model of herpes simplex encephalitis[J]. Antiviral Res, 2018, 149: 1-6. |