



2. 中国科学院昆明植物研究所植物化学与西部植物资源持续利用国家重点实验室, 云南 昆明 650201
2. State Key Laboratory of Phytochemistry and Plant Resources in West China, Kunming Institute of Botany, Chinese Academy of Sciences, Kunming 650201, China
阿片类药物是临床上用于治疗疼痛的最常见的药物。阿片类药物的靶点是阿片受体(opioid receptor,OR)[1],属于G蛋白偶联受体家族,分4种亚型,即μ受体、δ受体、κ受体和阿片样受体-1(ORL-1)。吗啡、芬太尼等传统阿片类镇痛药是μ受体选择性激动剂,广泛用于术后疼痛、慢性疼痛、癌痛、腹泻、咳嗽等的临床治疗[2]。然而,阿片类药物也有一系列的严重副作用,如成瘾、耐受、呼吸抑制、尿潴留等[3, 4],限制了其临床应用。
近期的研究已明确,不同OR的激动效应之间存在相互制衡,能产生增强镇痛、减弱副作用的效果。例如,联合用药研究发现,δ受体激动剂可减弱 μ受体激动引起的呼吸抑制、肌肉强直等副作用,而μ受体激动剂可减弱δ受体激动剂的惊厥副作用[5, 6];另一方面,κ受体激动剂能够减弱μ受体介导的镇痛耐受及成瘾性[7, 8]。因此,OR混合活性化合物有可能在保持镇痛活性的同时减轻阿片样副作用,近期受到广泛关注[9, 10, 11]。Neumeyer等[12, 13]发现κ/μ受体混合激动剂可降低成瘾性。Chang等[14, 15, 16, 17]发展了一类δ/μ受体混合激动剂,其中DPI-3290[15, 16, 17]同时具备κ受体激动活性,在强效镇痛的同时可降低呼吸抑制和躯体依赖副作用,在美国进入二期临床试验。
KUST201(又称DPI-125)[17]是与DPI-3290同期发现的另一δ/μ/κ受体三重激动剂,具备强效镇痛活性,副作用也明显轻微。本文报道我们近期通过大鼠模型实验对KUST201镇痛、耐受和戒断反应的研究结果。
1 材料与方法 1.1 材料SD大鼠购自成都达硕生物科技有限公司,♂,SPF级,体质量180~200 g,动物生产许可证号:SCXK(川)2013-24。盐酸纳洛酮、DMSO购自美国Sigma公司。KUST201、纳曲吲哚(NTI)分别按文献方法合成。夹尾镇痛实验使用W40190动脉钳[(上海医疗器械(集团)有限公司手术器械厂)]。热板镇痛实验使用YLS-6B型智能热板仪(济南益延科技发展有限公司)。KUST201溶液按如下方法配制:KUST201样品溶于适量DMSO(占总体积比例<5%)中,加入等当量浓盐酸后溶于生理盐水。
1.2 夹尾镇痛实验选取符合标准的大鼠,随机分组,每组8~10只。肌肉注射KUST201或空白对照(溶剂),将动脉钳夹在大鼠尾巴距顶部2.5 cm处,用计时器记录大鼠咬动脉钳或其它痛觉反应的时间,最长时间设为20 s,在给药前测定大鼠的基础痛阈,给药后20 min再次测定大鼠的痛阈,计算给药后的最大镇痛百分率(MPE%),计算公式如下:
MPE%=[(给药后痛阈-给药前痛阈)/(20-给药前痛阈)]×100%
1.3 热板镇痛实验选取符合标准的大鼠,随机分组,每组8~10只。肌肉注射KUST201或空白对照(溶剂),将大鼠放在(55±0.5)℃的金属恒温热板上,放置同时开启计时器,当大鼠出现舔后足或跳跃动作时,马上停止计时。为防止足部烫伤,观察截止时间设为60 s。在给药前测定大鼠的基础痛阈,给药后20 min再次测定大鼠的痛阈,计算给药后的最大镇痛百分率(MPE%),计算公式如下:
MPE%=[(给药后痛阈-给药前痛阈)/(60-给药前痛阈)]×100%
1.4 大鼠镇痛活性实验采用大鼠夹尾镇痛模型和热板镇痛模型测定痛阈变化,并使用NTI检测δ受体拮抗对镇痛的影响。将大鼠随机分组,测定基础痛阈后分别给予空白对照(溶剂)和KUST201(0.25~1.5 mg·kg-1,im)、NTI(2 mg·kg-1,sc)+KUST201(1.5 mg·kg-1,im,NTI给药20 min后注射)、NTI(2 mg·kg-1,sc),给药后每20 min测量大鼠的痛阈,计算MPE%。
1.5 大鼠慢性耐受实验采用大鼠夹尾镇痛模型和热板镇痛模型测定痛阈变化,并使用NTI检测δ受体拮抗对镇痛的影响。将大鼠随机分为3组,测定基础痛阈后分别给予KUST201(1.5 mg·kg-1,im)、NTI(2 mg·kg-1,sc) + KUST201(1.5 mg·kg-1,im,NTI给药20 min后注射)、NTI(2 mg·kg-1,sc),每天上午给药(9 am),测定给药后20 min的痛阈,计算MPE%。
1.6 大鼠戒断反应采用4 d递增法给予大鼠KUST201,然后用纳洛酮催促戒断。将大鼠随机分为4组,按梯度设定起始单次给药剂量(0.68、1.36、2.04、2.72 mg·kg-1,im),每天给药2次(8 am,4 pm),单次给药剂量按1倍、2倍、3倍逐日递增。d 4上午按3倍剂量给药3 h后注射纳洛酮(10 mg·kg-1,sc)。观察纳洛酮给药前后0.5 h的行为变化和给药前后1 h的体重变化。
1.7 统计学处理实验数据采用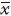 ±s表示,ED50值计算均采用GraphPad Prism软件,各组均数的比较采用单因素方差分析(one-way ANOVA),两组间比较采用t检验。
±s表示,ED50值计算均采用GraphPad Prism软件,各组均数的比较采用单因素方差分析(one-way ANOVA),两组间比较采用t检验。
KUST201和DPI-3290的大鼠夹尾和热板实验结果汇总如Fig1和Fig2。KUST201的夹尾镇痛在0.5 mg·kg-1剂量时达到最大镇痛(Fig1A),ED50为(0.34±0.15) mg·kg-1(Fig1B),热板镇痛在1.5 mg·kg-1时达到最大镇痛(Fig1C),ED50为(0.68±0.17) mg·kg-1(Fig1D)。作为对比,DPI-3290的夹尾镇痛在1.0 mg·kg-1时达到最大镇痛(Fig2A),ED50为(0.30±0.16) mg·kg-1(Fig2B),热板镇痛在4.0 mg·kg-1时达到最大镇痛(Fig2C),ED50为(2.55±0.93) mg·kg-1(Fig2D),与KUST201的镇痛活性类似。这一结果与已报道的研究结果[17]一致,即KUST201的镇痛活性与DPI-3290相当,优于吗啡。

|
| Fig.1 Antinociceptive effects of KUST201 on rats A: Time course of antinociceptive effects in tail-pinch test; B; Calculation of ED50 of tail-pinch test; C: Time course of antinociceptive effects in hot-plate test; D: Calculation of ED50 of hot-plate test. |

|
| Fig.2 Antinociceptive effects of DPI-3290 on rats A: Time course of antinociceptive effects in tail-pinch test; B: Calculation of ED50 of tail-pinch test; C: Time course of antinociceptive effects in hot-plate test; D: Calculation of ED50 of hot-plate test. |
由于KUST201的δ受体激动活性强于μ受体活性[17],为明确δ受体激活在KUST201镇痛中的作用,我们在镇痛时效实验中考查了δ拮抗剂纳曲吲哚(NTI)的影响(Fig3)。与NTI相比,KUST201组与KUST201+NTI组均产生明显的镇痛作用(P<0.01),热板实验中KUST201+NTI组的镇痛作用明显弱于KUST201组,差异具有统计学意义(P<0.01),表明KUST201的δ激动活性能协同增强经μ受体激动产生的镇痛效果。

|
| Fig.3 Effects of NTI on KUST201-induced antinociception in rats A: Tail-pinch test; B: Hot-plate test.**P<0.01 vs NTI;##P<0.01 vs NTI+KUST201 |
在大鼠的慢性耐受夹尾镇痛模型以及热板镇痛模型中,KUST201在前2 d均达到最大镇痛,但从d 3开始产生耐受,镇痛效果下降,到d 7时镇痛作用消失(Fig4)。Gengo等[17]报道了DPI-3290的慢性耐受夹尾镇痛模型实验,从d 2开始即产生耐受,到d 5镇痛作用消失。对比上述结果,KUST201的慢性耐受反应弱于DPI-3290。
与前述结果类似,NTI也能减弱KUST201的镇痛活性(P<0.01)(Fig4),表明在长期使用中,KUST201的δ激动活性也能增强μ受体激动的镇痛效果。

|
| Fig.4 Chronic tolerance of KUST201 in rats A: Tail-pinch test; B: Hot-plate test.**P<0.01 vs NTI;##P<0.01 vs NTI+KUST201 |
KUST201的躯体依赖性采用大鼠的纳洛酮催促戒断实验考察(Tab1)。戒断症状的评分标准如下:兴奋性、腹泻、流涎、舔生殖器、眼脸下垂等症状未出现时0分,出现时即给4分;体重减轻0~9 g时0分,10~15 g时4分,16~20 g时8分,21~25 g时12分,>25 g时18分;跳跃1~2次时2分,5~6次时4分,7~8次时6分,9~10次时8分,>10次时12分;扭体1~5次时2分,>5次时4分;前肢震颤、抓挠以及牙颤1~5次时2分,6~10次时4分,>10次时6分[18]。
| Sign | Initial dose of KUST201a and abstinence score/mg·kg-1 | |||
| 0.68 | 1.36 | 2.04 | 2.72 | |
| Average scoreb | 16±1.08 | 22.5±0.92 | 27±1.02 | 36±1.66 |
| Motor-related Signc | 10±1.05 | 15.5±0.94 | 19.8±1.01 | 27.2±1.36 |
| Non-motor-related Signd | 6±1.19 | 7±0.90 | 7.2±0.83 | 8.8±1.24 |
| Sign | Initial dose of DPI-3290b and abstinence score/mg·kg-1 | |||
| 0.2 | 1 | 2 | 5 | |
| Average scorec | 6.3±0.94 | 14±1.11 | 15±1.43 | 19±0.94 |
| Motor-related Signd | 6.3±0.87 | 13±1.39 | 13.7±1.82 | 18±1.16 |
| Non-motor-related Signe | 0 | 1±0.57 | 1.3±0.63 | 1±0.67 |
| a:Initial doses of KUST201 used are 2-,4-,6-,8-times of ED50 in tail-pinch test in rats; b:Intitial doses of DPI-3290 used are 0.67-,3.33-,6.67-,16.7-times of ED50 in tail-pinch test in rats; c:4-6 rats for each dose; d:Including jumping(escape attempts),wet-dog shake,head shake,forelimb tremor,digging,teeth chattering and writhing; e:Including irritability,diarrhea,salivation,licking penis,ptosis and weight loss in 3 h(≥ 10 g). | ||||
实验结果显示,KUST201和DPI-3290给药剂量的增加均引起大鼠戒断反应分值升高(Tab1,Fig5),但变化趋势有所不同。DPI-3290的戒断反应分值低于KUST201,最大分值(19±0.94,16.7×ED50)仅稍大于KUST201的起始分值(16±1.08,2×ED50),且运动无关症状明显减少(Tab1)。DPI-3290的剂量关系曲线上升平缓,而KUST201的曲线上升幅度较大(Fig5)。

|
| Fig.5 Naloxone-precipitated withdrawal abstinence scores in rats treated with KUST201 and DPI-3290 at the folds of tail-pinch ED50 |
值得注意的是,KUST201的戒断反应分值接近于μ受体激动剂(如吗啡,15~34,1~5×ED50)[17],但成瘾性明显降低。Traynor等[19]报道了猴自身给药模型中,KUST201与μ受体激动剂阿芬太尼的对比,结果表明二者镇痛活性相近,但 KUST201的成瘾倾向(ED50 2.5 μg·kg-1·inj-1)远弱于阿芬太尼(ED50 0.11 μg·kg-1·inj-1),最大响应仅为阿芬太尼的70%。
综合上述结果,KUST201的躯体依赖性高于DPI-3290,接近吗啡,但成瘾性低于阿芬太尼。由于KUST201和DPI-3290均为δ/μ/κ受体三重激动剂,KUST201的κ受体活性弱于DPI-3290[17],上述结果可能说明κ受体在各种副作用的调节中参与的程度有所不同,对戒断反应的调节作用弱于成瘾作用。
3 结论吗啡、芬太尼等传统阿片类镇痛药物尽管具有良好的镇痛效果,但伴有呼吸抑制、依赖性、成瘾性等严重副作用。我们的前期工作表明δ/μ/κ受体三重激动剂在呼吸系统安全性方面远优于传统阿片类镇痛药,是一类镇痛效果好、副作用少的新型阿片类药物[15]。本研究对δ/μ/κ受体三重激动剂KUST201的镇痛、耐受和戒断反应进行了研究。KUST201的夹尾镇痛ED50为(0.34±0.15) mg·kg-1,热板镇痛ED50为(0.68±0.17) mg·kg-1,镇痛作用可维持1 h,2 h后作用消失。慢性耐受实验中,KUST201从d 3开始产生镇痛耐受,d 7作用消失。δ拮抗剂NTI可部分抵消KUST201的镇痛作用,表明δ受体激动能增强μ受体激动的镇痛效果。戒断反应实验中,KUST201的剂量关系曲线在2~8倍夹尾镇痛ED50范围内上升明显。与已报道的δ/μ/κ受体三重激动剂DPI-3290相比,KUST201的镇痛活性相仿,镇痛耐受反应较弱,而戒断反应稍强。我们的结果为此类新型镇痛药物的进一步研究提供了实验依据。
| [1] | Feng Y,He X,Yang Y,et al. Current research on opioid receptor function[J]. Curr Drug Targets,2012,13(2): 230-46. |
| [2] | Al-Hasani R,Bruchas M R. Molecular mechanisms of opioid receptor-dependent signaling and behavior[J]. Anesthesiology,2011,115(6): 1363-81. |
| [3] | 沈 宁,郭瑞鲜,胡 芬,等. 脊髓Cx43通过JNK通路介导大鼠慢性吗啡镇痛耐受[J]. 中国药理学通报,2013,29(3): 372-6. Shen N,Guo R X,Hu F,et al. Spinal Cx43 mediates chronic morphine antinociceptive tolerance through JNK pathway in rats[J]. Chin Pharmacol Bull,2013,29(3):372-6. |
| [4] | 傅 璐,郭瑞鲜,莫利求,等. 脊髓c-Jun在NMDA受体NR2B亚基介导的大鼠吗啡镇痛耐受中的作用[J]. 中国药理学通报,2011,27(2): 248-52. Fu L,Guo R X,Mo L Q,et al. Role of c-Jun of spinal cord in NMDA receptor subunit NR2B mediated morphine antinociceptive tolerance in rats[J]. Chin Pharmacol Bull,2011,27(2): 248-52. |
| [5] | O′Neill S J,Collins M A,Pettit H O,et al. Antagonistic modulation between the delta opioid agonist BW373U86 and the mu opioid agonist fentanyl in mice[J]. J Pharmacol Exp Ther,1997,282(1):271-7. |
| [6] | Su Y F,McNutt R W,Chang K J. Delta-opioid ligands reverse alfentanil-induced respiratory depression but not antinociception[J]. J Pharmacol Exp Ther,1998,287(3): 815-23. |
| [7] | Wang Y H,Sun J F,Tao Y M,et al. The role of kappa-opioid receptor activation in mediating antinociception and addiction[J]. Acta Pharmacol Sin,2010,31(9):1065-70. |
| [8] | Pan Z Z. Mu-opposing actions of the kappa-opioid receptor[J]. Trends Pharmacol Sci,1998,19(3): 94-8. |
| [9] | 伊首璞,陈忠明,张继虹,等. 阿片类受体亚型间相互作用研究进展[J]. 中国药理学通报,2012,28(11): 1493-6. Yi S P,Chen Z M,Zhang J H,et al. Research progress of interactions among different opioid receptor subtypes[J]. Chin Pharmacol Bull,2012,28(11): 1493-6. |
| [10] | 沈 庆,刘慧芳,李 炜,等. μ/δ阿片受体相互调节作用及药物发现与设计策略[J]. 中国药理学通报,2010,26(1): 4-8. Shen Q,Liu H F,Li W,et al. Modulated interaction of μ/δ opioid receptors and the drug discovery and design strategy[J]. Chin Pharmacol Bull,2010,26(1): 4-8. |
| [11] | Fujii H. Twin and triplet drugs in opioid research[J]. Top Curr Chem,2011,299: 239-75. |
| [12] | Bowen C A,Negus S S,Zong R,et al. Effects of mixed-action kappa/mu opioids on cocaine self-administration and cocaine discrimination by rhesus monkeys[J]. Neuropsychopharmacology,2003,28(6):1125-39. |
| [13] | Zhang T Z,Yan Z H,Sromek A,et al. Aminothiazolomorphinans with mixed kappa and mu opioid activity[J]. J Med Chem,2011,54:1903-13. |
| [14] | Bishop M J,Garrido D M,Boswell G E,et al. 3-(αR)-α-[(2S,5R)-4-allyl-2,5-dimethyl-1- piperazinyl)-3-hydroxybenzyl]-N-alkyl-N-arylbenzamides: potent,non-peptidic agonists of both the mu and delta opioid receptors[J]. J Med Chem,2003,46(4): 623-33. |
| [15] | Gengo P J,Pettit H O,O′Neill S J,et al. DPI-3290[(+)-3-((α-R)-α-((2S,5R)-4-allyl-2,5- dimethyl-1-piperazinyl)-3-hydroxybenzyl)-N-(3-fluorophenyl)-N-methylbenzamide]. I. A mixed opioid agonist with potent antinociceptive activity[J]. J Pharmacol Exp Ther,2003,307(3): 1221-6. |
| [16] | Gengo P J,Pettit H O,O′Neill S J,et al. DPI-3290[(+)-3-((α-R)-α-((2S,5R)-4-allyl-2,5- dimethyl-1-piperazinyl)-3-hydroxybenzyl)-N-(3-fluorophenyl)-N-methylbenzamide]. II. A mixed opioid agonist with potent antinociceptive activity and limited effects on respiratory function[J]. J Pharmacol Exp Ther,2003,307(3):1227-33. |
| [17] | Gengo P J,Chang K J. Mixed opioid receptor agonists as a new class of agents for the treatment of moderate to severe pain[M]//Chang K J,Porreca F,Woods J H. The delta receptor. New York: Marcel Dekker,2004: 231-44. |
| [18] | Paul H K,Robert W M,Chang K J. A nonpeptidic delta opioid receptor agonist,BW373U86,attenuates the development and expression of morphine abstinence precipitated by naloxone in rat[J]. J Pharmacol Exp Ther,1993,267(2):883-7. |
| [19] | Traynor J R,Fantegrossi W,Woods J H. Evaluation of new compounds for opioid activity,annual report of CPDD drug evaluation program[J]. NIDA Res Monogr Ser, 2004,185: 131-59. |