



缺血性脑中风是一种发病率和死亡率都较高的脑血管病,阐明其发病机制、探索有效的防治措施一直是研究的热点。由活性氧(reactive oxygen species,ROS)引起的氧化应激是脑缺血/再灌注(ischemia reperfusion,I/R)损伤发生的重要机制之一。以往多数研究采用自由基清除剂或者天然抗氧化剂来减轻氧化损伤,但并不能产生理想的效果,近来越来越多的研究者关注ROS的来源。有报道表明,在脑缺血时ROS主要由黄嘌呤氧化酶产生,恢复血流再灌注时ROS则主要是由还原型烟酰胺腺嘌呤二核苷酸磷酸(nicotinamide adenine dinucleotide phosphate,NADPH)氧化酶产生[1]。因此,NADPH氧化酶成为研究如何减轻脑I/R损伤的热点。有报道表明,抑制NADPH氧化酶能够减轻脑I/R损伤,但具体的作用机制尚不明确[2]。
神经元是脑I/R损伤过程中最为敏感的细胞,它对I/R耐受性低。研究发现,脑I/R过程中往往伴随着神经元缝隙连接蛋白Cx36的蛋白表达异常[3]。进一步研究发现,蛋白激酶C(protein kinase C,PKC)可以磷酸化Cx36蛋白的特殊位点,从而改变缝隙连接通道的耦合[4]。有报道表明,改变Cx36的蛋白表达以及神经元缝隙连接通道的耦合能够明显减轻脑I/R损伤[3]。但是尚未有报道表明,抑制NADPH氧化酶对脑I/R损伤的保护作用是否与PKC激酶和Cx36蛋白表达有关。研究表明脑I/R过程中往往伴随着ROS生成增加,ROS的过度释放能够调控Bcl-2家族参与细胞凋亡[5, 6]。抑制NADPH氧化酶能够减少ROS的生成,那么,抑制NADPH氧化酶对脑I/R损伤的保护作用是否与调控Bcl-2家族参与凋亡有关?本文探讨抑制NADPH氧化酶保护脑I/R损伤的可能机制。
1 材料与方法 1.1 材料 1.1.1 实验动物SPF级♂ SD大鼠52只,体质量(300±20) g,安徽医科大学实验动物中心提供[动物许可证号:SCXK(皖)2011-002]。
1.1.2 主要试剂Apocynin和Cx36单克隆抗体购于Sigma公司;PKC单克隆抗体购于Abcam公司;Bax、Bcl-2单克隆抗体购自BOSTER公司;TTC(2,3,5,-氯三苯基四氮唑)、β-actin单克隆抗体、山羊抗小鼠IgG二抗、山羊抗兔IgG二抗均购于Sigma公司;其他试剂均为国产分析纯级。
1.2 实验方法 1.2.1 实验分组将52只大鼠随机分为4组:假手术组(sham组)、I/R组、I/R+apocynin组(I/R+Apo组)、I/R+apocynin+PKC抑制剂GF109203X组(I/R+Apo+GF组),每组13只。
1.2.2 模型制作大鼠称重,腹腔注射水合氯醛3 mL·kg-1麻醉,颈部正中切口,分离并结扎右侧颈总动脉、颈外动脉,在颈内动脉和颈外动脉分叉约1 mm处做一小切口,将直径0.32 mm的尼龙线插入颈总动脉,再经颈内动脉至大脑中动脉,进线约18~22 mm,栓塞大鼠大脑中动脉,造成局灶性脑缺血。Sham组进行上述操作,但不插入线栓阻断大脑中动脉血流。I/R组缺血2 h后,恢复脑血流再灌注24 h;I/R+Apo组于缺血前15 min尾静脉注射apocynin 2.5 mg·kg-1,其余操作同I/R组。I/R+Apo+GF组于缺血前15 min尾静脉注射apocynin 2.5 mg·kg-1,并于缺血15 min后尾静脉注射PKC抑制剂(GF109203X)80 μg·kg-1,其余操作同I/R组。
1.2.3 神经行为学评估根据文献报道的神经行为功能损伤的评分方法[7],对缺血2 h再灌注24 h后的大鼠进行神经功能评分(每组13只)。0分:无神经功能缺失症状,活动正常;1分:提尾时手术对侧的前肢内收,不能完全伸直;2分:行走时向手术对侧旋转;3分:行走时向手术对侧倾倒,行动不便;4分:不能自己行走或处于昏迷状态。神经行为评分≥1分为成功模型,评分采用双盲法,由未参与实验分组的成员完成。
1.2.4 脑梗死体积测定大鼠缺血2 h,再灌注24 h后快速断头取脑(每组6只),将大脑置于-20℃冰箱10 min。自额极2 mm向后连续等距切取6张冠状脑片,间距为2 mm。2% TTC避光染色30 min(37℃),正常脑组织染为红色,脑梗死组织染为白色。将染色好的脑片放置于4%多聚甲醛溶液中固定保存24 h,逐层拍照。拍照后用Image J软件测量脑片相应部位面积,计算脑梗死体积百分率。计算公式:脑梗死体积百分率/%=[对侧大脑皮质的面积-(同侧大脑皮质的面积-脑梗死的面积)]/对侧大脑皮质的面积×100%[8]。
1.2.5 HE染色法观察脑组织病理形态学变化将脑缺血2 h,再灌注24 h后的大鼠断头取脑(每组3只),将脑组织进行甲醛固定、石蜡包埋,冠状位切片,片厚5 μm,HE染色,封片,光学显微镜下观察脑组织形态学变化,采集图像。
1.2.6 Western blot检测蛋白表达变化将脑缺血2 h,再灌注24 h后的大鼠断头取脑(每组4只),夹取右侧大脑脑组织,提取蛋白,进行标准蛋白定量。于100℃加热5 min,每孔取20 μg蛋白样品经15% SDS-PAGE凝胶电泳分离后,以湿转法电转移至聚偏二氟乙烯(polyvinylidene fluoride,PVDF)膜上,用新鲜配制的含5%脱脂奶粉的磷酸盐缓冲液封闭2 h,洗膜,与Cx36(1.25 mg·L-1)、PKC(1 ∶1 000)、Bax(1 ∶2 000)、Bcl-2(1 ∶2 000)单克隆抗体共同孵育,4℃过夜,洗涤后山羊抗兔抗体(1 ∶5 000)摇床上室温孵育2 h,β-actin(1 ∶4 000)单克隆抗体室温孵育2 h即可,其二抗为山羊抗鼠抗体(1 ∶1 000)。二抗孵育完毕后,洗膜,将膜与曝光液充分接触。1 min后将膜放入凝胶图像分析系统进行拍照。曝光时间调整在5~10 min之间。采用Bio Imaging system(Gene Genius)对图像进行灰度扫描分析。
1.3 统计学分析采用SPSS 13.0软件分析实验结果,数据资料以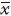 ± s表示,计量资料比较进行单因素ANOVA分析,统计图采用Sigma Plot 10.0绘制。
± s表示,计量资料比较进行单因素ANOVA分析,统计图采用Sigma Plot 10.0绘制。
脑I/R后,除sham组大鼠无神经功能缺失症状,其他各组大鼠都有不同程度的神经功能障碍。与sham组相比,I/R组大鼠神经行为学评分明显增高(P < 0.05)。与I/R组相比,I/R+Apo组大鼠的神经行为学评分明显降低(P < 0.01)。与I/R+Apo组相比,I/R+Apo+GF组大鼠的神经行为学评分则明显增高(P < 0.01)(Tab1)。
± s,n=13)
 |
0 | 1 | 2 | 3 | 4 | ± s |
| **P<0.01 vs I/R group; ##P<0.01 vs I/R+Apo group | ||||||
| Sham | 13 | 0 | 0 | 0 | 0 | 0 |
| I/R | 0 | 0 | 3 | 9 | 1 | 2.84±0.55 |
| I/R+Apo | 0 | 8 | 4 | 1 | 0 | 1.46±0.66 ** |
| I/R+Apo+GF | 0 | 3 | 6 | 4 | 0 | 2.08±0.75 ## |
如Fig1A结果显示,TTC染色后,大鼠大脑脑梗死区域呈白色,非梗死区域呈红色。梗死灶主要位于缺血侧大脑皮质、基底节和海马区域。用Image J软件测量脑片相应部位面积,计算脑梗死体积百分率。Sham组大鼠大脑无明显梗死,I/R组大鼠脑梗死明显,脑梗死体积百分率最高。与I/R组相比,I/R+Apo组大鼠大脑梗死体积明显减少,脑梗死体积百分率明显降低(P < 0.05)。与I/R+Apo组相比,I/R+Apo+GF组大鼠脑梗死体积百分率明显增加(P < 0.05)(Fig1B)。

|
|
Fig.1
Effect of inhibition of NADPH oxidase on the brain infarct volume of rats with cerebral I/R injury detected by TTC staining( ± s,n=6)
A: Slices of brain tissue,a:sham;b:I/R;c:I/R+Apo;d:I/R+Apo+GF; B: Quantitative analysis of brain infarct volume. *P<0.05 vs I/R group; #P<0.05 vs I/R+Apo group.
|
采用HE染色法观察大鼠脑组织病理形态学的变化。如Fig2所示,sham组大鼠脑组织中细胞排列整齐、均匀,细胞核圆形,细胞层次清晰;与sham组相比,I/R组大鼠脑组织中细胞的细胞核明显固缩,细胞层次混乱;与I/R组相比,I/R+Apo组大鼠脑组织中细胞的细胞核固缩有所减轻,细胞层次清晰;与I/R+Apo组相比,I/R+Apo+GF组大鼠脑组织病理形态学损伤明显增强。

|
| Fig.2 Effect of inhibition of NADPH oxidase on the pathological morphological changes of rats with cerebral I/R injury observed by HE staining A:Sham;B:I/R;C:I/R+Apo;D:I/R+Apo+GF |
采用Western blot检测Cx36、PKC以及凋亡相关蛋白Bax、Bcl-2的蛋白表达变化。与sham组相比,I/R组大鼠的Cx36、PKC蛋白表达明显降低(P < 0.05),但是Bax/Bcl-2的比值明显增高(P < 0.05)。与I/R组相比,I/R+Apo组大鼠的Cx36、PKC蛋白表达明显增高(P < 0.05),Bax/Bcl-2的比值则明显降低(P < 0.05)。与I/R+Apo组相比,I/R+Apo+GF组大鼠的Cx36、PKC蛋白表达明显降低(P < 0.05),但是Bax/Bcl-2的比值明显增高(P < 0.05) (Fig3、4)。

|
|
Fig.3
Effect of inhibition of NADPH oxidase on the expression of Cx36 and PKC of rats with cerebral I/R injury detected by Western blot( ± s,n=4)
A: Expression of Cx36 and PKC; B: Quantitative analysis of Cx36; C: Quantitative analysis of PKC. *P<0.05 vs sham group; #P<0.05 vs I/R group; △P<0.05 vs I/R+Apo group.
|

|
|
Fig.4
Effect of inhibition of NADPH oxidase on the expression of Bax and Bcl-2 protein of rats with cerebral I/R injury detected by Western blot( ± s,n=4)
A: Expression of Bax and Bcl-2 protein; B: Quantitative analysis of the Bax/Bcl-2 ratio. *P<0.05 vs sham group; #P<0.05 vs I/R group; △P<0.05 vs I/R+Apo group.
|
NADPH氧化酶广泛分布于大脑神经元细胞、星形胶质细胞、血管内皮细胞以及小胶质细胞,是脑缺血后,恢复血液再灌注时ROS最重要的来源。研究发现,使用NADPH氧化酶抑制剂或敲除NADPH氧化酶的基因可以明显减轻氧化损伤[9, 10],表明NADPH氧化酶在保护脑I/R损伤方面具有重要意义。Apocynin又叫夹竹桃麻素、罗布麻宁、香荚兰乙酮,是传统中医中的一种天然化合物。研究发现apocynin可以通过阻止p47 phox与NOX 2的组装从而抑制NADPH氧化酶的活化[11]。已有报道表明,apocynin能够减少大鼠脑梗死体积、神经功能缺陷以及细胞凋亡率,从而减轻脑I/R损伤,但具体的作用机制尚不明确[12]。
Cx36是缝隙连接蛋白家族的新成员,主要分布在脑组织以及视网膜。由Cx36组成的缝隙连接是最为常见的神经元细胞间偶联形式,是神经元电信号传导所必需的,而Cx36是唯一一个在神经系统中优先表达的缝隙连接蛋白,因此成为研究神经元缝隙连接功能改变的热点[13]。PKC激酶属于多功能丝氨酸/苏氨酸激酶,广泛调节各细胞的生理功能。有报道发现,PKC可以磷酸化Cx36蛋白的特殊位点,从而改变缝隙连接通道的耦合[4]。研究表明神经元之间的电耦合很少是静态的,动态变化的程度往往反映在蛋白表达水平的变化上[14]。有报道表明,在脑I/R过程中往往伴随着Cx36蛋白表达以及神经元缝隙连接功能异常,而改变Cx36蛋白表达以及神经元缝隙连接通道的耦合能够明显减轻脑I/R损伤[3]。在心脏以及肾脏组织中主要表达Cx43蛋白,而Src激酶可以使Cx43蛋白磷酸化或直接与Cx43蛋白相结合[15, 16]。有报道表明,NADPH氧化酶的活化可以通过Src激酶下调Cx43蛋白表达造成心肌损伤[15]。在肾病的动物模型中发现NADPH氧化酶的活化可以通过Src激酶上调Cx43蛋白表达,造成足细胞损伤[16]。但是尚未见报道表明在脑I/R过程中,NADPH氧化酶与PKC激酶和神经元缝隙连接蛋白Cx36之间的关系。
本实验结果表明,抑制NADPH氧化酶能够减少大鼠神经行为学评分,降低脑梗死百分率以及减轻脑组织病理形态损伤,从而减轻脑I/R损伤。为了探讨其可能的作用机制,我们采用Western blot检测Cx36、PKC蛋白表达变化。实验结果显示,与I/R组相比,使用NADPH氧化酶抑制剂apocynin后,明显增加大鼠Cx36、PKC的蛋白表达。在使用apocynin的基础上加用PKC抑制剂GF109203X,与单用apocynin相比,Cx36、PKC的蛋白表达明显降低。结果表明,抑制NADPH氧化酶减轻脑I/R损伤可能与增高Cx36、PKC的蛋白表达有关。
脑I/R过程中,ROS生成明显增加,ROS的过度释放可以调控Bcl-2家族参与细胞凋亡[5, 6]。Bcl-2家族能够调控线粒体膜通透性,在细胞的固有凋亡通路中有重要作用,被称为线粒体途径。脑缺血或缺氧后,线粒体膜蛋白的表达增加,促凋亡蛋白Bax从细胞质中进入线粒体,促进细胞色素C的释放。细胞色素C可以和 caspase-9前体的功能前区相互作用,导致caspase-3激活,并进一步激活下游的caspases[17](包括caspases-2、6、8、10),诱导细胞凋亡。抑凋亡蛋白Bcl-2位于线粒体壁,能够抑制细胞色素C的释放[17]。目前,许多学者用Bax/Bcl-2的比值作为判定细胞凋亡是否被抑制或促进的重要标志[18]。Western blot结果显示,使用NADPH氧化酶抑制剂apocynin后,与I/R组相比,能够降低Bax/Bcl-2的比值。在使用apocynin的基础上使用PKC抑制剂,与I/R+Apo相比,Bax/Bcl-2的比值则明显增高。
综上所述,抑制NADPH氧化酶能够减轻脑I/R损伤,并且能增高Cx36、PKC的蛋白表达,降低Bax与Bcl-2的比值。
| [1] | Abramor A Y, Scorziello A, Duchen M R, et al. Three distinct mechanisms generate oxygen free radicals in neurons and contribute to cell death during anoxia and reoxygenation[J]. J Neurosci, 2007, 27(5):1129-38. |
| [2] | 张朝弘,刘丹彦.NADPH氧化酶与脑缺血/再灌注损伤[J].国际病理科学与临床杂志, 2013, 33(3):246-50.Zhang Z H, Liu D Y. NADPH oxidase and cerebral ischemia/reperfusion injury[J]. Int J Pathol Clin Med, 2013, 33(3):246-50. |
| [3] | Belousov A B, Fontes J D. Neuronal gap junctions: making and breaking connections during development and injury [J]. Cell,2013, 36(4):227-36. |
| [4] | Kothmann W W, Li X, Burr G S, et al. Connexin 35/36 is phosphorylated at regulatory sites in the retina[J]. Vis Neurosci, 2007, 24(3): 363-75. |
| [5] | Sanderson T H, Reynolds C A, Kumar R, et al. Molecular mechanisms of ischemia-reperfusion injury in brain: pivotal role of the mitochondrial membrane potential in reactive oxygen species generation[J]. Mol Neurobiol, 2013, 47(1):9-23. |
| [6] | Li H, Wang L J, Qiu G F, et al. Apoptosis of HeLa cells induced by extract from Cremanthodium humile[J]. Food Chem Toxicol, 2007, 45(10):2040-6. |
| [7] | 雷军荣,秦 军,张 晶,等.姜黄素对大鼠缺血性脑损伤炎症反应和血脑屏障通透性的影响[J].中国药理学通报,2010, 26(1):120-3.Lei J R, Qin J, Zhang J, et al. Effects of curcumin on inflammatory reaction and blood-brain barrier permeability in rats following cerebral ischemic injury[J]. Chin Pharmacol Bull, 2010, 26(1):120-3. |
| [8] | Zhang Y M, Xu H, Sun H, et al. Electroacupuncture treatment improves neurological function associated with regulation of tight junction proteins in rats with cerebral ischemia reperfusion injury[J]. Evid Based Complement Alternat Med, 2014, 6(10):1-10. |
| [9] | Liu W, Chen Q, Liu J, et al. Normobaric hyperoxia protects the blood brain barrier through inhibiting Nox2 containing NADPH oxidase in ischemic stroke[J]. Med Gas Res, 2011, 1(1): 22. |
| [10] | Shen J, Bai X Y, Qin Y, et al. Interrupted reperfusion reduces the activation of NADPH oxidase after cerebral I/R injury[J]. Free Radical Bio Med, 2011, 50(12): 1780-6. |
| [11] | Stefanska J, Pawliczak R. Apocynin: molecular aptitudes[J]. Mediators Inflamm, 2008, 2008: 106507. |
| [12] | Pamenter M E, Ali S S, Tang Q, et al. An in vitro ischemic penumbral mimic perfusate increases NADPH oxidase-mediated superoxide production in cultured hippocampal neurons[J]. Brain Res, 2012, 1452(2): 165-72. |
| [13] | Palacios-Prado N, Hoge G, Marandykina A, et al. Intracellular magnesium-dependent modulation of gap junction channels formed by neuronal Connexin36[J]. J Neurosci, 2013, 33(11):4741-53. |
| [14] | Patel L S, Mitchell C K, Dubinsky W P, et al. Regulation of gap junction coupling through the neuronal connexin Cx35 by nitric oxide and cGMP[J]. Cell Commun Adhes, 2006, 13(1-2): 41-54. |
| [15] | Sovari A A, Rutledge C A, Jeong E M, et al. Mitochondria oxidative stress, Connexin43 remodeling, and sudden arrhythmic death[J]. Circ Arrhythm Eletrophysiol, 2013, 6(3):623-31. |
| [16] | Yan Q. NADPH oxidase-mediated upregulation of connexin43 contributes to podocyte injury[J]. Free Radic Biol Med, 2012, 53(6): 1286-9. |
| [17] | Kroemer G. Mitochondrial control of apoptosis: an introduction[J]. Biochem Biophys Res Commun, 2003, 304(3):433-5. |
| [18] | Huang X, Cho J K, Park S R, et al. GM-CSF inhibits apoptosis of neural cells via regulating the expression of apoptosis related proteins[J]. Neurosci Res, 2007, 58(1):50-7. |