



肝纤维化是持续性肝脏损伤的共同病理改变过程,主要表现为细胞外基质(extracellular matrix,ECM)在肝脏中的大量沉积,其持续发展可形成肝硬化和肝细胞癌[1]。肝纤维化的形成机制目前尚不清楚,研究表明肝星状细胞(hepatic stellate cell,HSC)的增殖活化是肝纤维化形成的关键,抑制HSC的增殖活化成为防治肝纤维化的重点[2]。有文献报道,Zeste基因增强子同源物2(enhancer of zeste homolog 2,EZH2)在纤维化形成中起重要作用[3]。肝纤维化形成过程中,MAPK/ERK和PI3K/AKT通路被激活并起重要作用。研究发现,EZH2能够激活MAPK/ERK和PI3K/AKT通路,从而引起疾病[4]。本研究拟应用EZH2的抑制剂DZNep和小干扰RNA(small interfering RNA,siRNA)抑制EZH2的表达,考察其对HSC增殖活化的调控作用,探究其调控机制,以期为肝纤维化的防治提供新的靶点。
1 材料 1.1 细胞株大鼠肝星状细胞株HSC-T6购于南京凯基生物科技发展有限公司。
1.2 试剂与仪器DZNep购于Selleckchem公司;TGF-β1购于 Peprotech公司;噻唑蓝MTT购于Sigma公司;DMSO购于Sigma公司;胰蛋白酶、DMEM培养基为Hyclone公司产品;胎牛血清购于杭州四季青公司;山羊抗小鼠IgG和山羊抗兔IgG购于北京中杉金桥生物技术有限公司;脂质体LipofectamineTM 2000、Opti-MEM购于Invitrogen公司;EZH2、p-ERK、ERK、p-AKT和AKT抗体购于Cell Signaling公司;α-SMA抗体购于北京博奥森生物技术有限公司;β-actin抗体购于北京中杉金桥生物技术有限公司;倒置荧光显微镜(OLYMPUS公司,日本);酶标仪(bio-tekEL)。
2 方法 2.1 HSC-T6细胞的培养应用含10% 胎牛血清的DMEM培养基,在37℃、5% CO2及饱和湿度条件下进行细胞培养,待细胞长至70%~80% 密度时进行细胞传代。
2.2 DZNep的应用加入DZNep 24 h前,将细胞传代至6孔培养板,每孔2×105细胞。实验分正常组、应用TGF-β1刺激的模型组、TGF-β1刺激并加入DZNep的恢复组。正常组不做任何处理;模型组加入TGF-β1刺激终浓度为10 μg · L-1,TGF-β1作用于细胞的时间为48 h;恢复组加入TGF-β1刺激终浓度为10 μg · L-1,并加入DZNep刺激终浓度为1 μmol · L-1,DZNep作用于细胞的时间为24 h。
2.3 siRNA的合成根据siRNA设计原则,设计针对EZH2基因的siRNA序列,在GenBank表达序列标签(EST)数据库应用BLAST检索,并确认所设计siRNA序列的唯一性,靶向目的基因EZH2的序列设计的两条链,正义链:5′-GGGCAUCUUUAUCAAAGAUTT-3′;反义链:5′- AUCUUUGAUAAAGAU GCCCTT-3′。同样方法构建阴性对照组的两条链,正义链:5′-UUCUCCGAACGUGUCACGUTT-3′;反义链:5′-ACGUGACACGUUCGGAGAATT-3′,与任何编码序列无同源性,由吉玛制药有限公司合成有效siRNA干粉制剂。
细胞培养及siRNA转染HSC-T6细胞培养于含10% 胎牛血清的DMEM培养基,37℃、5% CO2培养。转染24 h前,传代至6孔培养板,每孔2×105细胞。实验分正常组、模型组、阴性对照组和EZH2-siRNA实验组。正常组不做任何处理;模型组加入TGF-β1;阴性对照组转染FAM标记的阴性对照siRNA并加入TGF-β1;实验组转染EZH2-siRNA并加入TGF-β1。用Opti-MEM和脂质体作为转染试剂进行转染,使siRNA终浓度均为100 pmol · L-1,置于37℃、5% CO2培养箱转染6 h后,每组加入完全培养基含TGF-β1使刺激终浓度为10 μg · L-1,继续培养48 h,收获细胞。采用荧光倒置显微镜观察各组转染后的细胞形态并判断转染效果。
2.4 细胞增殖实验以MTT法检测HSC-T6细胞的增殖,分组同上。将培养的HSC-T6细胞以10% 胎牛血清的DMEM制备成1×108 · L-1的细胞悬液,接种于96孔板,每孔100 μL,过夜,待细胞贴壁后进行细胞转染,转染后6 h换液,每孔加入完全培养基200 μL含TGF-β1使刺激终浓度为10 μg · L-1,37℃、5% CO2培养箱中继续培养12、24、48、72 h,然后每孔加0.5% MTT贮存液20 μL继续孵育4 h,最后弃上清,每孔加入150 μL二甲基亚砜溶解细胞内结晶,置于恒温震荡器上震摇10 min,应用酶标仪于490 nm波长处测定吸光度值,以空白对照组作为对照组,其细胞存活率为100%,其余各组按:存活率/%=[(各实验组吸光度值-本底吸光度值)/(对照组吸光度值-本底吸光度值)]×100%。
2.5 Western blot检测EZH2、p-ERK、p-AKT、α-SMA蛋白表达细胞的培养、分组、处理同上。DZNep作用24 h或者转染EZH2-siRNA 48 h后,分别提取细胞总蛋白,应用BCA法测定蛋白浓度。应用10% SDS-PAGE凝胶进行电泳,在200 mA恒定电流下应用PVDF膜进行转膜,TBST洗膜后,使用5% 牛奶封闭非特异性结合位点,TBST洗膜后,在4℃于一抗(1 ∶500)中孵育过夜,TBST洗膜后,加入辣根过氧化物酶标记的山羊抗小鼠或抗兔二抗(1 ∶10 000)室温中孵育1 h,TBST洗膜后,加入ECL发光剂反应20 s,电脑显影成像,实验重复3次或以上。
2.6 统计学处理采用SPSS 13.0软件进行统计分析,数据以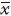 ± s表示。两组间比较采用t检验,多组间比较采用单因素方差分析。
± s表示。两组间比较采用t检验,多组间比较采用单因素方差分析。
加入TGF-β1的模型组EZH2蛋白表达水平明显升高,与正常组相比差异有显著性(P<0.01),加入TGF-β1和DZNep的恢复组EZH2蛋白表达水平明显降低,与模型组相比差异有显著性(P<0.01),见Fig1A;同时,模型组p-ERK和p-AKT蛋白表达水平明显升高,与正常组相比差异有显著性(P<0.01),恢复组p-ERK和p-AKT蛋白表达水平明显降低,与模型组相比差异有显著性(P<0.01),见Fig1B;与此同时,模型组α-SMA蛋白表达水平明显升高,与正常组相比差异有显著性(P<0.01),恢复组α-SMA蛋白表达水平明显降低,与模型组相比差异有显著性(P<0.01),见Fig1C。

|
| Fig 1 Protein expression of EZH2(A), p-ERK, p-AKT(B) and α-SMA(C) detected by Western blot after application of DZNep 1: normal group; 2: model group; 3: recovery group. **P<0.01 vs normal group; ##P<0.01 vs model group |
MTT实验结果显示,EZH2-siRNA瞬时转染对TGF-β1诱导的HSC-T6细胞的增殖有明显的抑制作用,EZH2-siRNA实验组作用12 h细胞抑制率与阴性对照组、模型组之间差异无显著性(P>0.05);EZH2-siRNA实验组作用24、48、72 h细胞抑制率明显高于阴性对照组、模型组(P<0.05)。同时,作用24、48、72 h时模型组、阴性对照组与正常组之间差异有显著性(P<0.05),见Fig2。

|
| Fig 2 Proliferation of HSC cells determined by MTT after transfection with siRNA of EZH2 1: normal group; 2: model group; 3: negative control group; 4: EZH2-siRNA experimental group. *P<0.05 vs normal group; #P<0.05 vs negative control group |
瞬时转染EZH2-siRNA 48 h后,EZH2蛋白表达明显降低,与阴性对照组相比差异有显著性(P<0.01),阴性对照组、模型组与正常组相比差异亦有显著性(P<0.01),见Fig3A;同时,EZH2-siRNA实验组p-ERK和p-AKT蛋白表达也明显降低,与阴性对照组相比差异有显著性(P<0.01),阴性对照组、模型组与正常组相比差异亦有显著性(P<0.01),见Fig3B;与此同时,EZH2-siRNA实验组α-SMA蛋白表达也明显降低,与阴性对照组相比差异有显著性(P<0.01),阴性对照组、模型组与正常组相比差异亦有显著性(P<0.01),见Fig3C。

|
| Fig 3 Protein expression of EZH2(A), p-ERK, p-AKT(B) and α-SMA(C) detected by Western blot after transfection with EZH2 siRNA 1: normal group; 2: model group; 3: negative control group; 4: EZH2-siRNA experimental group. **P<0.01 vs normal group; ##P<0.01 vs negative control group |
HSC的增殖活化是肝纤维化发生发展的关键环节,抑制HSC的增殖活化成为预防和治疗肝纤维化的重要手段之一[5]。近些年研究发现[6, 7, 8, 9],表观遗传学(包括DNA甲基化、microRNA和组蛋白修饰等)在肝纤维化的发生发展中扮演重要角色。组蛋白修饰是指在组蛋白相关酶的作用下,组蛋白发生乙酰化、磷酸化、甲基化和泛素化等修饰的过程。组蛋白的甲基化修饰是由组蛋白甲基转移酶来实现的,组蛋白的甲基化可发生在组蛋白的赖氨酸和精氨酸残基上,最终可导致基因的转录沉默或转录激活。研究发现[10],组蛋白的甲基化修饰与HSC的状态及转归密切相关。EZH2是一个重要的组蛋白甲基转移酶,通过特异性甲基化基因启动子区域组蛋白H3赖氨酸27,广泛参与了基因的转录沉默。在癌症的发生发展中,EZH2被认为是一个促癌基因,在子宫内膜癌、前列腺癌、肝癌等恶性肿瘤中高表达[11, 12, 13]。有研究发现,EZH2在肝纤维化形成中扮演重要角色,但是其具体机制尚不清楚。
有文献报道[14, 15],在生长因子TGF-β1等的刺激下,MAPK/ERK和PI3K/AKT信号通路被激活,进而影响到大鼠肝星状细胞HSC-T6的增殖活化。有研究发现,EZH2能够激活MAPK/ERK和PI3K/AKT信号通路从而引起疾病的发生。那么EZH2能否影响TGF-β1诱导的HSC的增殖活化,EZH2通过何种机制影响其增殖活化,目前尚未见文献报道。在该项研究中,应用TGF-β1诱导HSC增殖活化,同时应用EZH2的抑制剂DZNep和siRNA技术,抑制HSC-T6中EZH2的表达,观察EZH2表达下调后对TGF-β1诱导的HSC增殖活化的影响,及其对MAPK/ERK和PI3K/AKT信号通路的影响。应用EZH2的siRNA后,与阴性对照组比较,EZH2-siRNA实验组EZH2蛋白表达水平明显降低,表明瞬时转染实验成功。MTT法是评估细胞增殖的一种有效方法,MTT的结果表明,通过siRNA沉默EZH2的表达可明显抑制TGF-β1诱导的HSC-T6细胞的增殖。α-SMA是HSC活化的特征性标志,Western blot检测结果显示,应用抑制剂DZNep后,可明显降低α-SMA蛋白的表达;靶向干扰HSC-T6细胞中EZH2的表达,可明显降低α-SMA蛋白的表达。表明抑制EZH2的表达能影响TGF-β1诱导的HSC的增殖活化。结果进一步表明,应用TGF-β1刺激HSC-T6细胞可明显升高EZH2蛋白表达水平,也可明显升高p-ERK、p-AKT蛋白表达水平。应用抑制剂DZNep后可明显降低EZH2蛋白表达水平,也可明显降低p-ERK和p-AKT蛋白表达水平。靶向干扰HSC-T6细胞中EZH2基因的表达,可明显降低EZH2蛋白表达水平,也可降低p-ERK和p-AKT蛋白表达水平。表明抑制EZH2的表达能影响TGF-β1诱导的HSC中MAPK/ERK和PI3K/AKT信号通路的激活。抑制EZH2的表达能影响MAPK/ERK和PI3K/AKT信号通路的激活,是影响TGF-β1诱导的HSC增殖活化的机制。
综上,抑制HSC-T6细胞中EZH2基因的表达,能影响TGF-β1诱导的HSC中MAPK/ERK和PI3K/AKT信号通路的激活,从而抑制HSC的增殖活化。本研究从细胞分子水平阐明EZH2对HSC的增殖活化的调控作用及其可能的潜在调控机制。揭示了EZH2在肝纤维化发展中的重要作用,可能成为防治肝纤维化的潜在新靶点,还需要通过其它实验进一步验证。肝纤维化的形成过程非常复杂,EZH2如何调控基因的沉默,尚需进一步深入研究。
| [1] | Hernandez-Gea V, Friedman S L. Pathogenesis of liver fibrosis [J]. Annu Rev Pathol, 2011, 6:425-56. |
| [2] | Lee U E, Friedman S L. Mechanisms of hepatic fibrogenesis [J]. Best Pract Res Clin Gastroenterol, 2011, 25 (2):195-206. |
| [3] | Mann J, Chu D C, Maxwell A, et al. MeCP2 controls an epigenetic pathway that promotes myofibroblast transdifferentiation and fibrosis [J]. Gastroenterology, 2010, 138 (2):705-14. |
| [4] | Min J, Zaslavsky A, Fedele G, et al. An oncogene-tumor suppressor cascade drives metastatic prostate cancer by coordinately activating Ras and nuclear factor-κ B [J]. Nat Med, 2010, 16 (3):286-94. |
| [5] | Fallowfield J A. Therapeutic targets in liver fibrosis [J]. Am J Physiol Gastrointest Liver Physiol, 2011, 300 (5): G709-15. |
| [6] | 陶 辉, 黄 成, 杨晶晶, 等. RNAi介导MeCP2基因沉默对HSC-T6细胞活化增殖的影响[J]. 中国药理学通报, 2012, 28(3):333-6. Tao H, Huang C, Yang J J, et al. Experimental study of proliferation and activation of HSC-T6 cells through RNA inference targeting MeCP2 [J]. Chin Pharmacol Bull, 2012, 28(3):333-6. |
| [7] | Bian E B, Huang C, Wang H, et al. Repression of Smad7 mediated by DNMT1 determines hepatic stellate cell activation and liver fibrosis in rats [J]. Toxicol Lett, 2014, 224 (2):175-85. |
| [8] | He Y, Huang C, Sun X, et al. MicroRNA-146a modulates TGF-betal-induced hepatic stellate cell proliferation by targeting SMAD4 [J]. Cell Signal, 2012, 24 (10):1923-30. |
| [9] | Mannaerts I, Eysackers N, Onyema O O, et al. Class Ⅱ HDAC inhibition hampers hepatic stellate cell activation by induction of microRNA-29 [J]. PloS One, 2013, 8 (1):e55786. |
| [10] | Perugorria M J, Wilson C L, Zeybel M, et al. Histone methyltransferase ASH1 orchestrates fibrogenic gene transcription during myofibroblast transdifferentiation [J]. Hepatology, 2012, 56 (3):1129-39. |
| [11] | Jia N, Li Q, Tao X, et al. Enhancer of zeste homolog 2 is involved in the proliferation of endometrial carcinoma [J]. Oncol Lett, 2014, 8(5):2049-54. |
| [12] | Chinaranagari S, Sharma P, Chaudhary J. EZH2 dependent H3K27me3 is involved in epigenetic silencing of ID4 in prostate cancer [J]. Oncotarget, 2014, 5 (16):7172-82. |
| [13] | Au S L, Wong C C, Lee J M, et al. Enhancer of zeste homolog 2 epigenetically silences multiple tumor suppressor microRNAs to promote liver cancer metastasis [J]. Hepatology, 2012, 56 (2):622-31. |
| [14] | Bian E B, Huang C, Ma T T, et al. DNMT1-mediated PTEN hypermethylation confers hepatic stellate cell activation and liver fibrogenesis in rats [J]. Toxicol Appl Pharmacol, 2012, 264 (1):13-22. |
| [15] | Fang L, Zhan S, Huang C, et al. TRPM7 channel regulates PDGF-BB-induced proliferation of hepatic stellate cells via PI3K and ERK pathways [J]. Toxicol Appl Pharmacol, 2013, 272 (3):713-25. |