 2017, Vol. 37
2017, Vol. 37文章信息
- 孙鉴锋, 王建新.
- SUN Jian-feng, WANG Jian-xin.
- 一种基于E-index方法区分复杂性状的分析工具
- An Analysis Tool Based on E-index Method for Differentiating Complex Traits
- 中国生物工程杂志, 2017, 37(2): 93-100
- China Biotechnology, 2017, 37(2): 93-100
- http://dx.doi.org/DOI:10.13523/j.cb.20170214
-
文章历史
- 收稿日期: 2016-08-26
- 修回日期: 2016-09-03






2. 北京林业大学计算生物学中心 北京 100083
2. Center for Computational Biology, Beijing Forestry University, Beijing 100083, China
有效了解复杂性状的遗传调控方式,对农业、生物进化和生物医学等研究有着重要意义[1]。区分复杂性状是研究不同基因型作用机理的前提,也是开展生物多样性探究工作必不可少的步骤。国内外对于有效区分复杂性状的探索持续了数十年,已经在多个阶段取得了良好的成果与进展,但是目前的方法都不能很好地区分复杂性状。Functional Mapping (FM) 方法是一种通过选定数学参数,利用动态生长曲线加以区分复杂性状的方法,其区分方法对于过去各类QTL作图方法是一次重要的转变[2]。早期指标法 (earliness index,E-index) 在FM方法的基础之上进行了改进,是目前区分生物复杂性状极为有效的方法,这为进一步研究生物多样性提供了良好的策略[3]。不难发现,在生物遗传学和生物多样性的研究上面诞生了众多方法[4],但是支撑这些新方法和新理论的技术与工具有一定的滞后性。此外,在当前信息范式和信息技术的不断推动下,数据可视化技术[5]逐渐渗透到生物学领域,生物数据可视化成为现代生物学的热点研究话题[6],并在生物多样性、生物医学和基因组学等研究领域应用广泛[7]。在这样的背景下,为了加快生物工作者们对于区分复杂性状的研究步伐,本文将E-index方法转变为一个基于互联网模式驱动的实用工具,为生物工作者们提供了更加便利的研究方式。
1 区分复杂性状概述 1.1 QTL定位与检测Geldermann为了研究基因如何调控性状,提出了控制数量性状的数量性状位点 (quantitative trait loci,QTL) 的概念[8],是进一步分析生物基因型的有效途径,也为下阶段了解复杂性状的遗传模式和交互作用奠定了良好的基础。随着各项生物基因组计划的进行,QTL检测与定位成为现代生物遗传学的一项重要研究内容,对基因表达分析的巨大潜能促进了遗传基因组学的发展[9]。进入20世纪80年代,分子标记技术得到了良好的发展,并应用于生物遗传学领域,是从基因和DNA分子层面上研究遗传性状的有力工具。单标记分子法[10]是最早用来进行QTL定位的分子标记方法,利用部分的分子标记连锁图谱结合分子标记的均值来分析基因型,在早期的QTL定位研究中应用颇为广泛。但是,该方法的估计结果只能说明QTL与标记相关,不能分析QTL的遗传效应,更不能确定与多个QTL之间连锁效应,而且观测样本量较大,降低了其检测效率。
随后,Lander等[11]对QTL定位的作图方法做了细致研究,区间定位方法的成功应用改善了过去QTL的检测模式,并极大地提高了检测效率和检测准确性。但是区间作图法不能有效评价出QTL的效应,甚至会导致QTL定位不准。为了更加精细的QTL作图,结合线性回归理论的复合区间作图法 (CIM) 被用于分析QTL遗传效应[12]。然而,CIM方法依赖于模型预设参数,对同一区间设置不同的协因子导致的QTL效应检测结果差异很大,每次甄别最佳协因子也会带来较大成本。后来,Kao等[13]提出一种多区间作图方法 (MIM),利用极大似然法和逐步选择变量的方式推导出标记基因的QTL主效应,但是对非QTL主效应的检测并不灵敏。一般而言,QTL检测往往发生在生物个体或组织器官生长成熟的一段时期 (某种表型较为显著),关注更多时期的数据有待于进一步尝试。这也促使研究者们产生了一个想法:使用更多时期甚至整个生长周期中的数据区分生物个体或组织器官的基因型[14]。
1.2 复杂性状一般而言,复杂性状指由多基因和非遗传因素共同作用的一类性状,更加侧重遗传和环境因素共同交织作用的一类性状,但是复杂性状的分析必须建立在对QTL理论基础之上进行分析[15]。例如,小鼠体重的性状由数十个基因进行调控。过去的研究者们会将复杂性状的多个基因进行逐一拆分,单独进行研究,但是此类做法会带来较高的时间成本和研究难度。可以看到,传统的QTL定位以及检测方法,主要侧重于成熟后的表型 (有限的数据),而非整个动态生长过程。无论是区间作图法,还是复合区间作图方法来完成QTL定位,都是建立在对部分数据 (生物个体或组织器官具有一定成熟度时) 分析的基础之上设计的方法,其本质上是一种统计策略的进步。众所周知,生物个体的数量性状受到基因的调控,同时也可能受到诸多环境因素的影响。传统QTL检测手段忽略了这个过程,从某种意义而言,遗传工作者需要关注到更多富有生物学意义的数据,才能更好地探究生物个体的复杂性状。
在已有的各类QTL作图方法的基础上,Wu等[16]结合了统计学和基因组学的知识,设计出了更加强大的分析策略,称为Functional Mapping (FM)。FM方法运用函数的思想,来体现出生物个体和组织器官的动态生长过程。该方法是对生物动态的复杂性状研究领域的一种成功尝试,具有里程碑式的意义。生长函数对应的曲线能够细微地体现出生物个体或组织器官在不同时期生长的姿态等特性,这些数据能够更加形象生动地和富含生物学意义地,表现出整个生物个体或组织器官的生长轨迹。
FM方法主要通过调节Logistic函数的不同参数值,来模拟不同生物个体或组织器官的生长过程,并通过最大似然估计法和最大期望算法估算出QTL的遗传效应等相关信息。它在很大程度上推进了动态区分生物复杂性状的进程,但是并不适应于全部的生物个体或组织器官的生长过程。例如,在经典的Scammon生长模式中[17],归纳出了人类组织器官生长发育的4类生长曲线,而FM方法并不能完全描述其中所有类型的生长曲线。
E-index方法克服了FM方法不能描述所有的生物生长曲线的缺点和不足,通过获得生物生长过程中的离散数据点 (原始的复杂性状数据),采用三次样条曲线拟合数据点的方式[18],可以较为真实和准确地还原出任意复杂性状 (针对某种表型,如高度或体重等) 的生长曲线。E-index是建立在统计分析学基础之上的方法和框架,有一定的理论体系结构支撑,使用积分的思想捕捉到生物生长过程中某种性状增加或是减少的变化值,最后会使用T检验[19]完成整个区分过程。
1.3 区分复杂性状工具的现状近年来,随着生物信息化进程的不断推动,生物工作者们不仅需要便利的分析软件和工具,还需要丰富多样的信息展现形式[20]。对于定位和识别生物基因的工具已有一些成功的案例,例如QTL IciMapping软件,提供了多类QTL作图方法,并且支持数据类型多,对结果提供了十分丰富的展现形式,功能全面且完善[21]。然而,现有的区分复杂性状的工具依然较少,不能对已有方法和理论形成良性循环和支撑。对于定位基因的各类方法和理论,国内外普遍的做法是通过多个软件合作的方式,进行数据计算,过程记录与结果转换。这使得区分性状和识别基因的过程需要花费生物工作者们较多的时间和精力,在一定程度上降低了工作效率。E-index理论涉及多类数学学科,其计算复杂度较高,直接将理论投入生产实践当中,具有一定的难度。本文设计并实现了基于E-index方法的分析工具 (E-index Application,EIA),是少有的采用生长曲线区分复杂性状的分析工具,在理论与实践的转换之间提供了一座桥梁,便于使用者理解E-index方法,从而增强了E-index方法的可用性,易用性和高能性。
2 EIA分析工具设计 2.1 E-index理论E-index方法成功克服了FM方法不能描述所有的生物生长曲线的不足,提供了更加多层次,全面化区分复杂性状的方式,并利用数理知识和统计学框架,能够对所有类型生长曲线进行区分,并且区分效果显著。但是E-index方法涉及的计算过程较为复杂,完成一次E-index的计算和绘图通常要借助多个软件和工具编写程序等手段,这为推广和应用E-index带来了极大挑战。EIA分析工具搭载了全套的E-index理论体系,检验过程和数据可视化。使用者无需再关注计算过程,只需要集中目光设计相应的实验,EIA分析工具就能自动完成整个复杂性状的区分过程。与此同时,还能将生物动态生长的整个过程用数据可视化技术表现出来。
EIA分析工具通过获取两种生物个体或者组织器官等样本数据进行分析。这里可以假设具有基因型A的样本观测矩阵为:
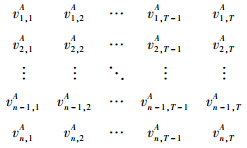 |
(1) |
基因型为B的样本观测矩阵为:
 |
(2) |
将上述基因型为A和B的样本观测矩阵中的每一个值随同相应的时间输入至各自的文件挡中,上传文件并且提交分析即可完成动态绘图。
为了更清楚地展现过程变量,假定基因型A的E-index值为EA,基因型B的E-index值为EB,分别代表了A、B基因型的生物某类性状的平均早期适应度。分析工具在自动执行相应的算法后,不仅会给出各组样本的E-index值,还会给出样本的均值、方差、标准差等基本统计量,以便让使用者多角度地和全方位地了解区分复杂性状的过程。
2.2 检验区分效果EIA分析工具计算E-index值后,将会采用T-test检验方式对控制两种相对性状的基因进行检验和区分,并给出假设检验的结果,以此完成整个生物个体或组织器官基因型的区分过程。为此,假设:
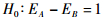 |
(3) |
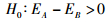 |
(4) |
其中H0为假设两种表型 (性状) 之间的差异不显著,H1为备择假设。若假设这两种表型之间的差异并不显著,可认为具备相同基因型的生物个体或组织器官。若拒绝H0,接受H1,即两种表型之间的差异显著,可认为二者是具有不同基因型的生物个体和组织器官。
2.3 EIA分析工具的设计方案自生物信息数据库海量增长后,以B/S结构[22]为支撑的各类系统成为主流的生物应用类工具[23]。EIA分析工具是基于web平台开发出来的应用软件,使用者需要处于互联网环境,通过浏览器与EIA分析工具交互。EIA分析工具通过B/S (Browser/Server) 结构模式来实现,其网络拓扑结构如图 1所示。

|
| 图 1 EIA分析工具的网络拓扑结构 Figure 1 Network topology of EIA analysis tool |
用户在PC端使用浏览器与集成了EIA分析工具的特定服务器交互,当服务器端接收到用户的指令,按照用户的请求返回分析结果,以此达到交互的目的。EIA分析工具的设计目标能够满足一般情况的复杂性状分析工作,以及实时绘图等工作需要。特别是为使绘图环节能够具有较高的用户体验度,EIA分析工具搭载了Highcharts数据可视化方案作为动态绘图的引擎。Highcharts在当前生物信息学领域主要用于生物海量数据库的数据提取,数据操作等方面,其绘图可以直接保存为PDF,这对于快速分析区间数据特征提供了极大的便利[24]。
EIA分析工具技术方案用图 2所示的流程图来表示,共分为5部分。第一步是获取数据阶段,其数据格式有一定要求。数据应包含该生物或组织器官等各个生长时间的数据,以及描述性状的数据 (比如高度,体重等),数据文件中涵盖了具有某种基因型的生物个体或组织器官的全部观测数据,可以写入该基因型下的多个样本个体的观测数据,将复杂性状数据随观测时间依次间隔排列,提交至EIA分析工具。第二步,对各组数据进行三次样条插值,将三次样条函数表达式作为生长函数。第三步,利用数据可视化技术实时并且动态地绘制各组数据对应的不同生物的生长曲线。第四步,可以根据输入进来的不同组别的样本观测值,计算出相应的E-index的值。最后,利用统计学方法T-test法进行假设检验,得到最终的区分结果。

|
| 图 2 EIA分析工具集成E-index的技术方案实现流程 Figure 2 Implementing flows of technical scheme of EIA analysis tool integrated E-index |
随着数据可视化技术的飞速发展,可视化界面除了显示图形本身之外,还能够提供与图形相关的各类信息。这些信息可以是生长曲线的基因型,观测样本的序列号,以及各个观测点下的具体数值等,可以帮助使用者在分析过程中了解当前环节的具体推进情况,甚至对各个出错的环节进行纠正。根据E-index的理论方案,完成EIA分析工具,其主界面为图 3所示 (详细链接:https://github.com/GenomicsGenuine/E-index-Application-Analysis-Tool)。

EIA分析工具中提供的绘制生长曲线的部分包含有如下功能:(1) 能够捕捉到不同生物个体和组织器官的基因类型,并标记不同的颜色加以区分;(2) 能够动态地画出生长曲线,并且获取生长曲线的表达式;(3) 能够动态地、有选择性地去除或是隐藏不需要的曲线,逐一地观查部分的曲线。
三次样条函数方法具有连续和光滑的函数特性,能够较好地弥补出多个数据点之间缺失的信息,EIA分析工具通过三次样条曲线基本上能够绘制以及复原任意一个生物个体或组织器官的复杂生长轨迹。此外,EIA分析工具上提供了全套的操作流程和相关说明等信息 (如图 4所示),将用户使用的过程变得简洁易懂、可操作性强、体验度好,同时为广大生物研究者和爱好者们在区分生物复杂性状等上面提供了相应的导读。

|
| 图 4 EIA分析工具使用说明界面 Figure 4 The description view for usage of EIA analysis tool |
利用计算机仿真实验的方法,本文对scammon生长模式下的神经组织型 (反J型曲线) 和4组生殖组织型 (J型曲线) 两种类型组织动态的区分复杂性状。这里,复杂性状可以定义为器官或组织随时间变化的生长程度。计算机仿真模拟生成4组神经组织型和4组生殖组织型的数据。导入数据之后,由分析工具自动完成绘图 (如图 5所示),曲线的类型为三次样条函数曲线。

|
| 图 5 EIA分析工具绘制生长曲线红色代表生殖组织型的生长曲线;绿色曲线是神经组织型生长曲线 Figure 5 Drawing a growth curves by EIA analysis tool The red curve is the growth curve of the reproductive tissue. The green curve is the nerve tissue growth curve |
表 1是EIA分析工具的输出结果。可以看到由于t值显著大于t0.01(6)=3.143,所以,假设检验结果为红色曲线的生物个体或组织器官 (即生殖组织型) 与绿色曲线的生物个体或组织器官 (即神经组织型) 分别来自不同基因型,也就是说,红色曲线与绿色曲线所代表的性状是由不同基因型调控的。
| 类型 | 样本 | E | t | |||
| 1 | 2 | 3 | 4 | |||
| 神经组织 | 0.297 | 0.292 | 0.301 | 0.303 | 0.298 | 50.983 |
| 生殖组织 | 0.611 | 0.628 | 0.640 | 0.629 | 0.627 | |
5 展望 5.1 EIA分析工具的未来发展
生物复杂性状包含了多维度的具有生物学意义的信息[25],如何采用更加丰富的方式和方法来全面表达信息将是未来对于复杂性状研究的热点。E-index方法是从一维角度出发来设计区分复杂性状的方法,而E-index方法更进一步的研究将建立在多维信息化模型上,这将更为全面地找出不同曲线代表的复杂性状的深层次含义和信息。EIA分析工具将在这个角度上扮演着重要的角色,不断地辅助和加快E-index理论的发展。
5.2 生长曲线应用前景生长曲线在饲料工业和微生物加工业中的生产控制环节扮演着重要角色。这些行业的生产过程依赖生长曲线分析生物特征,EIA分析工具的实时和自动地绘制生长曲线的用途,将对其分析工作有所帮助。例如,研究菌类生物的生长曲线可以找到最佳的增殖 (死亡) 时间段,从而可以控制该时间段的养殖方式,达到生产控制的目的。再如,枯草芽孢杆菌是一种用于猪粪除臭的菌株[26],了解其生长周期能够合理地制定方案,加大其繁殖效率,减少臭味气体的排放,净化人们赖以生存的生态环境。
此外,生长曲线在生物的基因工程研究[27]中应用广泛。测定酵母菌生长繁殖的最佳时期,利用基因工程的手段,将高效的酿酒酵母基因与其重组,能够极大地提高酵母菌的生产效率,对于生态制剂产业有很大的帮助[28]。此外,利用生长曲线分析微生物的生长周期,定位复杂性状疾病的基因[29],可以大力推动现代医学和药剂学的发展。
5.3 生物数据可视化随着生物各领域陆续进入互联网的发展潮流,海量的信息与数据随之而来,生物大数据时代正式兴起。大数据时代中的数据意味着"更多"、"更杂",但是"更好"[30]。这里的"更好"不仅是过去探讨的数据之间的因果关系,更多地体现在挖掘出数据背后的多种关联性,找寻和挖掘出数据的本质[31],这都将依赖于数据的多元化展现形式。可以看到,信息和数据从过去的书面化表示方式变为电子化的表示方式,生物工作者们对数据的诉求也变得更加强烈,不仅希望看到数据本体,也希望看到更加直观的数据表现形式,生物数据可视化便是处理这些海量数据的优先选择[32]。生物数据可视化是生物信息学[33]、基因组学[34]和神经信息学[35]等领域必不可少的研究手段,借助于数据可视化技术,这些领域能够生动地、直观地和全面地挖掘出生物信息,为现代生物研究注入了新鲜血液。生物数据可视化才刚刚兴起,在未来大数据驱动下的计算生物研究中拥有广阔的发展前景。
6 结束语E-index方法解决了区分复杂性状的难题,成功地应对了现代生物遗传学的挑战。E-index方法的实质是利用数理统计分析的手段,对数据含义进行深层次的挖掘,这切合了当前生物大数据的发展路线。E-index存有一定的改进空间,其改进将是对生物大数据维度更广的,层次更深的数据和信息挖掘。
在信息科技飞速发展的今天,EIA分析工具应运而生,其快速地进行成果转换的做法迎合未来生物信息化发展方向。EIA分析工具建立在最新的研究成果E-index方法的基础上,自动地、实时地和高效地完成区分复杂性状过程,是一次将理论快速转换为生产实践成果的成功尝试。EIA分析工具将数据可视化技术成功地应用到区分复杂性状当中,但是目前的可视化表现形式较为单一,还需要更多功能上的扩充,以应对未来生物大数据的挑战。与时俱进,在未来探究生物多样性的道路上,需要更多的实用性技术和工具的参与,才能不断加快生物遗传学和基因组学的发展。
| [1] | Sousa S A, Leităo J H, Martins R C, et al. Bioinformatics applications in life sciences and technologies. Biomed Research International, 2016, 2016(1) : 1–2. |
| [2] | Wu R, Lin M. Functional mapping——how to map and study the genetic architecture of dynamic complex traits. Nature Reviews Genetics, 2006, 7(3) : 229–237. DOI:10.1038/nrg1804 |
| [3] | Qi J, Sun J, Wang J. E-index for differentiating complex dynamic traits. Biomed Research International, 2015, 2016(1) : 1–13. |
| [4] | 陈英, 黄敏仁, 王明麻. 植物遗传转化新技术和新方法. 中国生物工程杂志, 2005, 25(9) : 94–98. Chen Y, Huang M R, Wang M M. The application of new technology and methods in plant transformation. China Biotechnology, 2005, 25(9) : 94–98. |
| [5] | Yu G, Wang L G, He Q Y. ChIP seeker:an R/Bioconductor package for ChIP peak annotation, comparison and visualization. Bioinformatics, 2015, 31(14) : 2382–2383. DOI:10.1093/bioinformatics/btv145 |
| [6] | Kannan S. Multi-dimensional, multi-configuration compilation phase output visualization technique. Journal of Computing in Civil Engineering, 2016, 23(6) : 363–371. |
| [7] | Che D, Wang H. GIV:A tool for genomic islands visualization. Bioinformation, 2013, 9(17) : 879–882. DOI:10.6026/bioinformation |
| [8] | Geldermann H. Investigations on inheritance of quantitative characters in animals by gene markers Ⅰ. Methods Tag Theoretical & Applied Genetics Theoretische Und Angewandte Genetik, 1975, 46(7) : 319–330. |
| [9] | Jansen R C, Nap J P. Genetical genomics:the added value from segregation. Trends in Genetics, 2001, 17(7) : 388–391. DOI:10.1016/S0168-9525(01)02310-1 |
| [10] | Martinez M L, Vukasinovic N, Freeman G A. Random model approach for QTL mapping in half-sib families. Genetics Selection Evolution, 1999, 31(4) : 319–340. DOI:10.1186/1297-9686-31-4-319 |
| [11] | Lander E S, Botstein D. Mapping Mendelian factors underlying quantitative traits using RFLP linkage maps. Genetics, 1989, 121(1) : 185–199. |
| [12] | Jansen R C. Interval mapping of multiple quantitative trait loci. Genetics, 1993, 135(1) : 205–211. |
| [13] | Kao C H, Zeng Z B, Teasdale R D. Multiple interval mapping for quantitative trait loci. Genetics, 1999, 152(3) : 1203–1216. |
| [14] | Zhao W, Chen Y Q, Casella G, et al. A non-stationary model for functional mapping of complex traits. Bioinformatics, 2005, 21(10) : 2469–2477. DOI:10.1093/bioinformatics/bti382 |
| [15] | Rieseberg L H, Sinervo B, Linder C R, et al. Role of gene interactions in hybrid speciation:evidence from ancient and experimental hybrids. Science, 1996, 272(5262) : 741–745. DOI:10.1126/science.272.5262.741 |
| [16] | Wu R, Ma C X, Casella G. Functional mapping of quantitative trait loci underlying the character process:a theoretical framework. Genetics, 2002, 161(4) : 1751–1762. |
| [17] | Morihara N. An anthropological study on somatometrical characters with growth of the head and face of the Bunun tribe in Taiwan aborigines. Journal of the Kyushu Dental Society, 1988, 42 : 559–579. |
| [18] | James R M, Panico V D. Evaluation of drag integral using cubic splines. Journal of Aircraft, 2015, 11(8) : 494–496. |
| [19] | Zimmerman D W. Comparative power of student T test and Mann-Whitney U Test for unequal sample size and variances. Journal of Experimental Education, 2014, 55(3) : 171–174. |
| [20] | Lawlor B, Walsh P. Engineering bioinformatics:building reliability, performance and productivity into bioinformatics software. Bioengineered, 2015, 6(4) : 193–203. DOI:10.1080/21655979.2015.1050162 |
| [21] | Lei M, Li H, Zhang L, et al. QTL Ici mapping:integrated software for genetic linkage map construction and quantitative trait locus mapping in biparental populations. Crop Journal, 2015, 121(3) : 269–283. |
| [22] | Hu B, Deng C, Ye J. Design and implementation of visual electronic commerce based on browser/server architecture. Advanced Materials Research, 2011, 271-273 : 336–339. DOI:10.4028/www.scientific.net/AMR.271-273 |
| [23] | 孙清鹏. 生物信息学应用教程. 北京: 中国林业出版社, 2012. Sun Q P. Bioinformatics Application Tutorial. Beijing: China Forestry Publication, 2012. |
| [24] | Dan T, Léger S, Belliveau L, et al. Metabo Hunter:an automatic approach for identification of metabolites from 1 H-NMR spectra of complex mixtures. Bmc Bioinformatics, 2011, 12(1) : 1–22. DOI:10.1186/1471-2105-12-1 |
| [25] | Komatsu H, Iguchi T, Ueda M, et al. Clinical and biological significance of transcription termination factor, RNA polymerase Ⅰ in human liver hepatocellular carcinoma. Oncology Reports, 2016, 35(4) : 2073–2080. |
| [26] | 高颖, 褚维伟, 张霞, 等. 猪粪除臭微生物筛选及其生长曲线测定. 山地农业生物学报, 2011, 30(1) : 47–51. Gao Y, Zhu W W, Zhang X, et al. Isolation and determination of growth curve of deoderizing microorganism from swine manure. Journal of Mountain Agriculture and Biology, 2011, 30(1) : 47–51. |
| [27] | Bailey B, Izarra A, Alvarez R, et al. Cardiac stem cell genetic engineering using the αMHC promoter. Regenerative Medicine, 2016, 4(6) : 823–833. |
| [28] | 郭春叶, 龚月生, 刘林丽, 等. 酵母菌生长曲线的测定及其转葡萄芪合酶基因重组菌遗传稳定性的检测. 安徽农业科学, 2007, 35(7) : 1909–1910. Guo C Y, Gong Y S, Liu L L, et al. Measurement of yeast growth curve and genetic stability examination of recombinant yeast transferred grape stilbene synthase (STS). Journal of Anhui Agricultural Sciences, 2007, 35(7) : 1909–1910. |
| [29] | Yu C H, Pal L R, Moult J. Consensus genome-wide expression quantitative trait loci and their relationship with human complex trait disease. Omics A Journal of Integrative Biology, 2016, 20(7) : 400–414. DOI:10.1089/omi.2016.0063 |
| [30] | 李志刚, 朱志军. 大数据时代:生活、工作与思维的大变革. 浙江: 浙江人民出版社, 2013. Li Z G, Zhu Z J. Big Data Age:A Revolution That Will Transform How We Live, Work, and Think. Zhejiang: Zhejiang People Press, 2013. |
| [31] | 尤元海, 张建中. 基因表达谱芯片的数据挖掘. 中国生物工程杂志, 2009, 29(10) : 87–91. You Y H, Zhang J Z. Data mining from microarray gene expression profile. China Biotechnology, 2009, 29(10) : 87–91. |
| [32] | Kyoda K, Tohsato Y, Ho K H, et al. Biological dynamics markup language (BDML):an open format for representing quantitative biological dynamics data. Bioinformatics, 2015, 31(7) : 1044–1052. DOI:10.1093/bioinformatics/btu767 |
| [33] | Kadoch C, Hargreaves D C, Hodges C, et al. Proteomic and bioinformatic analysis of mammalian SWI/SNF complexes identifies extensive roles in human malignancy. Nature Genetics, 2013, 45(6) : 592–601. DOI:10.1038/ng.2628 |
| [34] | Luo W, Brouwer C. Pathview:an R/Bioconductor package for pathway-based data integration and visualization. Bioinformatics, 2013, 29(14) : 1830–1831. DOI:10.1093/bioinformatics/btt285 |
| [35] | Salman A, Malony A, Turovets S, et al. Concurrency in electrical neuroinformatics:parallel computation for studying the volume conduction of brain electrical fields in human head tissues. Concurrency & Computation Practice & Experience, 2015, 28(7) : 2213–2236. |