 2017, Vol. 37
2017, Vol. 37文章信息
- 王曦光, 王娟, 张琳.
- WANG Xi-guang, WANG Juan, ZHANG Lin.
- 拟南芥蛋白质丰度与基因翻译效率关联分析
- A. thaliana Protein Abundance Analysis Coresponding with Elongation Efficiency
- 中国生物工程杂志, 2017, 37(2): 40-47
- China Biotechnology, 2017, 37(2): 40-47
- http://dx.doi.org/DOI:10.13523/j.cb.20170207
-
文章历史
- 收稿日期: 2016-06-20
- 修回日期: 2016-08-17






2. 中国农业科学院农业资源与农业区划研究所 北京 100081
2. Agricultural Resources and Regional Planning Institute, Chinese Academy of Agricultural Science, Beijing 100081, China
蛋白质是构成生物体的基本物质,是生命活动的主要承担者,它与生物体的各种生命活动和表征迹象息息相关。蛋白质的合成在包括原核生物和真核生物等所有生物体内都是一个精确调控的复杂的生理过程。蛋白质的合成具有动态性、复杂性、时间和空间上的不确定性,生物体各个部分蛋白质表达水平是不同的,在不同生命阶段及不同环境条件下,蛋白质表达水平也是不同的。蛋白质组学的研究提供了大量蛋白质丰度的数据,目前的研究一方面从mRNA丰度、蛋白质合成降解速度来研究蛋白质丰度的变化,另一方面从基因的特征来研究蛋白质丰度的变化[1]。Marquerat等[2]和Schwanhäusser等[3]分别对酵母和NIH3T3细胞进行研究时发现,mRNA丰度和蛋白质丰度具有较强的相关性,但在高等生物中该相关性逐渐减弱。Vogel等[4]对髓母瘤细胞的研究结果显示序列特征与蛋白质丰度变化也有一定的相关性, Ingvarsson[5]和McHardy等[6]研究表明密码子使用偏性与蛋白质丰度分布规律有密切关系。
大量实验数据和研究结果表明,绝大多数的蛋白质表达具有规律性,某一种特定基因在该生物体内的表达量和丰度值往往具有典型的分布规律和可预估的范围。蛋白质丰度受到基因表达各个过程的影响,其中翻译阶段的影响作用相对较大,它直接影响蛋白质的表达量,翻译的主要过程包括翻译起始、翻译延长和翻译终止三个阶段。目前的研究并没有从基因表达的角度对蛋白质丰度的变化做出很好的解释,而蛋白质的丰度却正是基因表达的最终衡量指标。通过对基因序列特征及相应蛋白质丰度关联性分析,可以比较准确地判断基因序列特征对蛋白质合成效率影响的程度[7-10]。
密码子是核酸序列携带信息和蛋白质序列携带信息间对应的基本规律,是生物体内信息传递的重要环节之一。64种遗传密码子在绝大多数生物体中的使用频率都是不一样的,即编码同一种氨基酸的密码子在不同物种中的使用频率不同。密码子偏好的现象在动物和植物中都非常普遍,它是由基因突变、遗传漂变及自然选择三者共同作用下产生的,最终形成了该物种在翻译效率和翻译精准性之间的平衡。自然选择的作用导致了密码子偏好和基因表达效率之间的紧密联系,即高表达基因具有明显的密码子使用偏好,所以密码子的使用偏好是衡量蛋白质翻译延伸效率的重要反应指标之一[11-14]。那些被频繁利用的密码子称为最佳密码子,而不经常利用的密码子称为稀有密码子或低利用率密码子。Sharp等[14](1987) 引入密码子适应指数CAI (codon adaptation index) 来反应蛋白质的翻译延伸效率,其计算基于高表达基因中不同密码子的使用频率[15-16]。随着生物信息学研究的深入,目前的研究显示CAI有一定的局限性,它没有考虑背景核苷酸突变所造成基因编码序列中核苷酸的偏差[16-18]。Xia[19]提出采用翻译延伸指数ITE(translation elongation index) 来衡量蛋白质翻译延伸效率。ITE的计算将密码子按照第三位分成了两组,分别以嘌呤结尾 (R-ending) 和嘧啶结尾 (Y-ending),从而避免了因基因组的突变 (背景核苷酸不同频率) 造成的密码子使用偏倚。
本研究借助于拟南芥蛋白质组学的大量实验数据并结合最新生物信息学研究成果对拟南芥的蛋白质丰度和基因特征进行了深入分析,其研究结果有助于揭示拟南芥蛋白质组的整体分布,理解蛋白质丰度和基因表达之间的关系,帮助寻找不同组织中拟南芥基因组中的共表达基因,并为系统生物学和拟南芥的生物学建模提供一定基础数据。
1 材料和方法 1.1 蛋白质丰度数据来源拟南芥 (Arabidopsis thaliana),又名阿拉伯芥、鼠耳芥、阿拉伯草,是植物学、遗传学和农业科学研究中最重要的模式生物之一。国内外对拟南芥的研究数据相对都比较多,本研究中拟南芥的蛋白质丰度数据主要来自pax-db (Protein Abundance Across Organisms) 网站。该网站是由生物信息学和系统生物学组开发,由瑞士信息研究所 (SIB) 资助在苏黎世大学发起的系统生物学研究项目。Pax-db是一个综合的蛋白丰度查询数据库,其中包含的蛋白质丰度信息覆盖到生物体的各个不同的组织器官[20-21]。在Pax-db中的蛋白质丰度实验数据是公开和可用的,本研究的蛋白质丰度主要来源于参考文献[22-23]中的实验数据。
1.2 拟南芥基因序列来源拟南芥的基因测序从2000年左右开始,目前全部序列的测序已经完成。拟南芥共有5个染色体,3万多条基因序列[24-29]。其中每个染色体所对应的基因序号,以及蛋白质数量、基因数量等详细数据如表 1所示,本研究中的序列来源于NCBI (美国国家生物技术信息中心, http://www.ncbi.nlm.nih.gov/) 的GenBank核酸数据库。拟南芥具有基因组小的特点,非常有利于生物信息学和分子生物学的研究[30-31]。
| 染色体 | RefSeq号 | 蛋白质数量 | rRNA数量 | tRNA数量 | 其他RNA数量 | 基因个数 | 失效基因个数 |
| 1 | NC_003070.9 | 9 263 | - | 240 | 218 | 8 433 | 924 |
| 2 | NC_003071.7 | 5 560 | 2 | 96 | 149 | 5 513 | 1 043 |
| 3 | NC_003074.8 | 6 908 | 2 | 93 | 134 | 6 730 | 1 080 |
| 4 | NC_003075.7 | 5 356 | - | 79 | 116 | 5 140 | 832 |
| 5 | NC_003076.8 | 8 089 | - | 123 | 127 | 7 507 | 948 |
| MT | NC_001284.2 | 117 | 3 | 21 | - | 131 | - |
| Pltd | NC_000932.1 | 85 | 7 | 37 | - | 129 | - |
1.3 拟南芥蛋白质丰度分析
从pax-db下载后的蛋白质丰度按不同组织器官进行分类,共有花、叶、幼叶、根茎、种子、幼苗、心皮、果实、花芽和花粉共计10种不同的组织器官,并在不同组织分组进行蛋白质丰度值的排序。选取每个组织中丰度较高和丰度较低的50种蛋白质并记录。统计每种高丰度和低丰度蛋白质在各个不同组织中出现的次数,并按蛋白质出现的次数再次从高到低排序。所得到的是拟南芥在各种不同组织中,蛋白质丰度都处于较高值和较低值的蛋白质。
1.4 蛋白质翻译延伸效率指数计算拟南芥的CAI值由CodonW计算得出。首先将拟南芥的GeneBank序列合成为一个整合的gb格式文件,然后将其蛋白质编码部分序列提取出来转换为fas格式,并输入到CodonW中。在计算时,首先通过选项4计算其COA值,再通过引用计算所得的cai.coa计算其最后的CAI值。需要注意的是,CAI的值不是绝对的,采用不同的参考集合会得到不同的CAI值[15-16]。
ITE是由DAMBE计算得出,ITE的计算基于高表达基因中密码子的使用频率及低表达基因中密码子的使用频率的差异,同时将密码子按照第三位碱基的不同分为嘌呤结尾密码子和嘧啶结尾密码子。与CAI值类似,ITE也是一个相对参考值,所得到的具体结果和参考集合 (高表达基因密码子使用频率及低表达基因密码子使用频率) 有关。高表达的基因应该比低表达基因具有高的ITE值。ITE的计算方式如公式1所示,其中Fi代表密码子i的使用频率,Ns为意义密码子的个数[16-17, 19]:
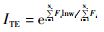 |
(1) |
本研究中所用到的系统分析方法主要有t检验、方差齐性检验、wilcox检验、线性回归分析和pearson相关分析,所使用的工具主要有R语言统计分析软件包和office Excel 2013。其中数据存储和整理主要用Excel 2013,数据处理和分析主要用R语言。本研究的技术路线图如图 1所示。

|
| 图 1 本研究技术路线图 Figure 1 Research roadmap |
(1) 生物信息学软件DAMBE (data analysis in molecular biology and evolution),是渥太华大学生物系夏旭华教授编写的序列综合分析软件,它包含了多种常用的生物信息学算法用于核苷酸序列和氨基酸序列的分析,在描述性基因组学、比较基因组学和分析进化研究中具有非常重要的地位,本研究所使用的版本为5.5.25 [18, 32-33]。
(2) CodonW,是一个专门用来计算密码子使用的开源软件分析包,目前最新版本为1.4.2,也是本研究中CAI计算所使用的工具①。
① http://codonw.sourceforge.net/Tutorial.html.
(3) R语言软件包,是由新西兰奥克兰大学的Ross Ihaka和Robert Gentleman开发的,是一套开源的集数据操作、计算和图形展示功能整合的软件包,本研究中所使用的版本为3.1.2。
2 结果与分析 2.1 高丰度和低丰度蛋白质从NCBI上下载拟南芥的全部染色体的所有基因,去除无效基因后共得到35 377条有效基因序列。按不同组织以蛋白质的丰度值进行排序,并选取其排名在前50的基因,进行整理分析得到拟南芥在10个不同组织中都具有高丰度的蛋白质、基因序列号及对应的组织器官,如表 2所示,其中在5个以上组织具有高丰度的蛋白质共计30种。在3个以上组织都是低表达的基因有20种,在5个组织以上都是低表达的基因只有2种。
| 基因ID | 根 (root) |
种子 (seed) |
幼苗 (shoot) |
心皮 (carpel) |
子叶 (cotyledon) |
花 (flower) |
花芽 (flower bud) |
幼叶 (juvenile leave) |
叶 (leaf) |
花粉 (pollen) |
合计 (totally) |
| ATCG00490 | - | Y | Y | Y | Y | Y | Y | Y | Y | - | 8 |
| AT1G07920 | Y | Y | Y | Y | - | - | Y | - | Y | Y | 7 |
| AT1G07930 | Y | Y | Y | Y | - | - | Y | - | Y | Y | 7 |
| AT1G07940 | Y | Y | Y | Y | - | - | Y | - | Y | Y | 7 |
| AT5G60390 | Y | Y | Y | Y | - | - | Y | - | Y | Y | 7 |
| ATCG00680 | - | - | Y | Y | Y | Y | Y | Y | Y | - | 7 |
| AT1G56070 | Y | Y | - | Y | - | Y | Y | - | - | Y | 6 |
| AT1G02780 | Y | - | Y | Y | - | Y | Y | - | - | Y | 6 |
| AT2G39730 | - | - | Y | Y | Y | - | Y | Y | Y | - | 6 |
| ATCG00120 | - | - | Y | - | Y | Y | Y | Y | Y | - | 6 |
| AT1G67090 | - | - | Y | - | Y | Y | Y | Y | Y | - | 6 |
| ATCG00270 | - | - | Y | Y | Y | - | Y | Y | Y | - | 6 |
| AT4G38970 | - | - | Y | - | Y | Y | Y | Y | Y | - | 6 |
| AT3G60750 | - | - | Y | - | Y | Y | Y | Y | Y | - | 6 |
| AT2G36530 | Y | - | - | Y | - | Y | Y | - | - | Y | 5 |
| AT1G07660 | Y | - | Y | Y | - | Y | Y | - | - | - | 5 |
| AT1G07820 | Y | - | Y | Y | - | Y | Y | - | - | - | 5 |
| AT2G28740 | Y | - | Y | Y | - | Y | Y | - | - | - | 5 |
| AT3G45930 | Y | - | Y | Y | - | Y | Y | - | - | - | 5 |
| AT3G46320 | Y | - | Y | Y | - | Y | Y | - | - | - | 5 |
| AT3G53730 | Y | - | Y | Y | - | Y | Y | - | - | - | 5 |
| AT5G59690 | Y | - | Y | Y | - | Y | Y | - | - | - | 5 |
| AT5G59970 | Y | - | Y | Y | - | Y | Y | - | - | - | 5 |
| AT3G26520 | Y | - | Y | - | Y | - | Y | Y | - | - | 5 |
| AT5G17920 | Y | - | - | - | - | Y | Y | Y | - | Y | 5 |
| ATCG00480 | - | - | Y | - | Y | - | Y | Y | Y | - | 5 |
| AT1G06680 | - | - | Y | - | Y | Y | - | Y | Y | - | 5 |
| AT1G20340 | - | - | Y | - | Y | Y | - | Y | Y | - | 5 |
| AT4G10340 | - | - | Y | - | Y | - | Y | Y | Y | - | 5 |
| AT5G35630 | - | - | Y | - | Y | - | Y | Y | Y | - | 5 |
| Y表示基因在该组织中具有高丰度值 (排名在前50),-表示该基因在该组织中不具有较高丰度值 (排名不在前50) | |||||||||||
在高表达基因中,在7个组织中具有高丰度的蛋白质共有5个,分别为AT1G07920,AT1G07930,AT1G07940,AT5G60390,ATCG00680。其中ATCG00680(蛋白质名称为PSBB) 结合叶绿素并有助于促进PSⅡ核心复合物的初级光诱导光化学过程,与光合作用密切相关。其他四个基因是构成翻译延长因子1(Elongation factor 1) 的核心蛋白质,可以促进GTP依赖性氨酰tRNA结合于A位的核糖体,为细胞执行核心功能,是蛋白质合成的必需蛋白。与能量代谢及物质转运密切相关的基因有很多也具有高表达性,如核酮糖二磷酸羧化酶大亚基 (ATCG00490) 和小亚基 (AT1G67090),它们主要用于催化1,5-二磷酸核酮糖和CO2生成二分子甘-3-磷酸甘油酸反应,其中大亚基蛋白质在8个组织中都处于高丰度。其次还有很多和叶绿体作用相关的基因,包括谷氨酰胺合成酶 (AT5G35630),氧化酶激酶 (AT2G39730),ATP合成酶α、β亚基 (ATCG00120,ATCG00480),放氧增强蛋白 (AT1G06680),叶绿素结合蛋白 (AT4G10340),光系统Ⅱ D2蛋白,醛缩酶 (AT4G38970),转酮酶 (AT3G60750) 等多种蛋白质,它们和拟南芥光合作用和能量转换有着密切关系。除以上基因外,与DNA复制及转录以及氨基酸和蛋白质合成的有关的基因有很多也是高表达基因,例如,双功能的烯醇化酶2(AT2G36530)、半胱氨酸甲基1(AT5G17920),60s亚基L19-1蛋白质 (AT1G02780) 等。此外,还有水通道蛋白TIP1-2(AT3G26520),它主要参与植物在高渗环境下渗透压的调节。由此可见,很多高表达基因都有共表达的特性,它们在一起才可以高效地执行细胞功能的整个过程。
从不同组织来看,花芽与幼苗的蛋白质组成比较接近,其次是花芽与心皮,花芽与花。在采用聚类分析之后,可以发现花粉与其他组织器官的蛋白质表达存在明显差异,幼叶与子叶的蛋白表达、花芽与花的蛋白表达最接近。和整体综合之后的表达最为接近的组织为根茎组织,其次是心皮。此外,从低表达的列表中可以看出,蛋白种类与高表达有明显不同,高表达蛋白很多为结构蛋白而低表达的蛋白质多为调控蛋白 (表 3)。
| 基因ID | 根 (root) |
种子 (seed) |
幼苗 (shoot) |
心皮 (carpel) |
子叶 (cotyledon) |
花 (flower) |
花芽 (flowerbud) |
幼叶 (juvenile leave) |
叶 (leaf) |
花粉 (pollen) |
合计 (totally) |
| AT3G63430 | Y | - | Y | Y | Y | - | - | - | Y | - | 5 |
| AT4G02660 | - | Y | Y | - | Y | - | Y | Y | - | - | 5 |
| AT2G13680 | Y | Y | - | - | - | - | Y | - | - | - | 3 |
| AT3G48190 | Y | - | - | - | - | - | - | - | Y | Y | 3 |
| AT1G68330 | Y | - | Y | - | - | - | - | - | Y | - | 3 |
| AT3G50380 | - | Y | Y | Y | - | - | - | - | - | - | 3 |
| AT4G36080 | - | Y | - | Y | Y | - | - | - | - | - | 3 |
| AT2G48060 | - | Y | - | - | - | - | Y | - | - | Y | 3 |
| AT2G40030 | - | - | Y | - | - | - | - | Y | Y | - | 3 |
| AT2G42270 | - | - | Y | - | Y | - | - | Y | - | - | 3 |
| AT1G77460 | - | - | Y | - | Y | - | - | - | - | Y | 3 |
| AT4G03030 | - | - | Y | - | Y | - | - | - | Y | - | 3 |
| AT3G47940 | - | - | Y | - | Y | - | - | - | Y | - | 3 |
| AT3G28770 | - | - | Y | Y | Y | - | - | - | - | - | 3 |
| AT5G60040 | - | - | - | Y | Y | - | - | Y | - | - | 3 |
| AT1G03060 | - | - | - | Y | Y | - | - | Y | - | - | 3 |
| AT2G10256 | - | - | - | Y | - | Y | Y | - | - | - | 3 |
| AT5G48310 | - | - | - | - | Y | Y | - | - | - | Y | 3 |
| AT1G08600 | - | - | - | - | - | Y | Y | Y | - | - | 3 |
| Y表示基因在该组织中具有低丰度值 (排名在前50),-表示该基因在该组织中不具有较低丰度值 (排名不在前50) | |||||||||||
2.2 高表达低表达基因CAI值与ITE值比较
如图 2a和2b所示,高表达基因的ITE值高于低表达基因的ITE值 (平均值分别为0.74和0.68),但CAI值的差别不明显,在低表达基因组CAI平均值为0.72略高于高表达基因组的CAI平均值。在高表达基因中ITE的值明显高于对应的CAI值,但在低表达基因中ITE值与CAI差别不大。将高表达和低表达两组CAI值和ITE值分别做t检验,p值分别为1.80481E-25和3.43681E-61,都具有明显差别。

|
| 图 2 基因的CAI值与ITE值比较图 Figure 2 Gene CAI value and ITE value (a) High expression gene (b) Low expression gene |
ITE与CAI都是用来衡量蛋白质延伸效率的指标,但ITE的计算不仅考虑了高表达基因中密码子的使用频率,而且兼顾了由于核苷酸突变导致的整个基因组中核苷酸的偏差问题 (将同一个密码子家庭分成嘌呤结尾和嘧啶结尾的不同组)。在编码同一个氨基酸的不同密码子家庭中,密码子的第三个差异 (嘌呤或嘧啶) 决定了运输其编码氨基酸的转运RNA (tRNA) 对相应密码子的翻译效率。ITE有效地解决了CAI的这种局限性,可以更加准确地描述翻译延长效率对蛋白质丰度的有效贡献度[17-18, 30]。
2.3 高表达与低表达基因长度比较基因长度这里指的是蛋白质编码区域的核苷酸序列长度。目前的研究认为,肽链的合成速度由翻译起始效率和翻译延长效率共同决定。真核细胞的翻译起始效率主要由Kozak序列的强弱决定,而翻译的延长效率则由密码子偏好和序列长度共同起作用。假设肽链在延长的时候使用相同的密码子的延长效率大致相等,可以很容易理解序列长度较短且结构相对简单的蛋白质合成的速度更快,效率更高[34-36]。本研究中,将蛋白质丰度统计分析后得到拟南芥的高丰度基因255个和低丰度基因369个。两组基因长度先进行方差齐性检验 (F=0.0603, p < 2.2e-16), 说明两组数据的方差不齐,选用Wilcox检验结果显示,两组基因的长度有显著性差异 (结果W=4512.5, p-value < 2.2e-16,),高表达的基因平均长度明显短于低表达的基因,其平均值分别为893和3 596。
这一结果也合理地解释了蛋白质在合成和细胞的物质及能量利用方面的经济性和高效性,较短的序列长度所生成的蛋白质结构也相对简单。这一结果与其他研究者[37-39]在研究HeLa细胞的转录组和蛋白质组时的研究结果相符,结构蛋白相对调控蛋白序列较短,同时结构蛋白相对调控蛋白丰度较高[29-31]。
2.4 蛋白质丰度与ITE相关性分析将拟南芥的全部组织蛋白质丰度值 (整合后的数据) 和ITE值相对应,共得到19 506条有效数据。将蛋白质丰度值进行对数转换并和相应基因的ITE值进行pearson相关性分析。分析结果显示,蛋白质丰度与ITE值有较强的相关性 (R=0.3635,p < 2.2e-16)。线性回归方程为y=0.0041x + 0.6863,其中x为ITE,y为对数转换后的蛋白质丰度,如图 3所示。该回归模型显示ITE与蛋白质丰度有较强的正相关性,在拟南芥的蛋白质丰度变化中,ITE可以提供13.2%(R2=0.132) 的解释,这和Xia[35]对E.coli的研究结果具有一致性。

|
| 图 3 蛋白质丰度对数转换后与ITE线性回归 Figure 3 Proten logarithmic abundance and ITE linear regression |
生物体内蛋白质组数据和丰度值是动态变化的,分析蛋白质丰度的变化情况及所受到的影响因素是非常重要的研究方向。本文通过近年来对拟南芥的蛋白质组数据和基因信息整合和统计分析,从生物信息学的角度对拟南芥各组织中蛋白质丰度变化规律和基因翻译效率进行了深入的研究。
研究结果表明,在拟南芥的基因中有多种蛋白质在不同组织中都处于高丰度值。高丰度的蛋白质主要执行细胞核心功能,是维持细胞基本生物机能必需蛋白, 主要涉及到蛋白质合成、能量代谢等功能,并且多个高表达基因具有共表达的特性。和低表达基因相比,高表达基因的编码序列较短,结构相对简单,且密码子使用偏性更高。在翻译延伸效率的研究中发现,拟南芥的CAI值无法有效地解释蛋白质丰度的整体变化,而ITE和蛋白质丰度的变化有很强的正相关性并可以提供约13%左右的解释,能够预测拟合拟南芥蛋白质丰度值的变化。
| [1] | 李定辰, 杨冬, 姜颖, 等. 蛋白质丰度调控及整体分布的规律性认识. 中国科学生命科学, 2013, 43(1) : 54–62. Li D C, Yang D, Jiang Y, et al. Insights into the regular patterns of protein abundance regulation and distribution. Scientia Sinica Vitae, 2013, 43(1) : 54–62. DOI:10.1360/052012-368 |
| [2] | Marquerat S, Schmidt A, Codlin S, et al. Quantitative analysis of fission yeast transcriptomes and proteomes in proliferating and quiescent cells. Cell, 2012, 151(3) : 671–683. DOI:10.1016/j.cell.2012.09.019 |
| [3] | Schwanhausser B, Weiss M, Simonovic M, et al. Global quantification of mammalian gene expression control. Nature, 2011, 473(7347) : 337–342. DOI:10.1038/nature10098 |
| [4] | Vogel C, Abreu R S, Ko D, et al. Sequence signatures and mRNA concentration can explain two-thirds of protein abundance variation in a human cell line. Mol Syst Biol, 2010, 6(1) : 400. |
| [5] | Ingvarsson P K. Gene expression and protein length influence codon usage and rates of sequence evolution in Populus tremula. Mol Biol Evol, 2007, 24(3) : 836–884. |
| [6] | McHardy A C, Puhler A, Kalinowski J, et al. Comparing expression level-dependent features in codon usage with protein abundance:an analysis of 'predictive proteomics'. Proteomics, 2004, 4(1) : 46–45. DOI:10.1002/(ISSN)1615-9861 |
| [7] | Ikemura T.Correlation between codon usage and tRNA content in microorganisms. In:Hatfield D L, Lee B J, Pirtle R M. Transfer RNA in Protein Synthesis, Boca Raton:CRC Press, 1992:87-111. |
| [8] | Na D, Lee D. RBSDesigner:software for designing synthetic ribosome binding sites that yields a desired level of protein expression. Bioinformatics, 2010, 26(20) : 2633–2634. DOI:10.1093/bioinformatics/btq458 |
| [9] | Nakamoto T. A unified view of the initiation of protein synthesis. Biochem Biophys Res Commun, 2006, 341(3) : 675–678. DOI:10.1016/j.bbrc.2006.01.019 |
| [10] | Schattner P, Brooks A N, Lowe T M. The tRNAscan-SE, snoscan and snoGPS web servers for the detection of tRNAs and snoRNAs. Nucleic Acids Res, 2005, 33 : 686–689. DOI:10.1093/nar/gki366 |
| [11] | Seo S W, Yang J S, Kim I, et al. Predictive design of mRNA translation initiation region to control prokaryotic translation efficiency. Metab Eng, 2012, 15(1) : 67–74. |
| [12] | Tuller T, Waldman Y Y, Kupiec M, et al. Translation efficiency is determined by both codon bias and folding energy. Proc Natl Acad Sci U S A, 2010, 107(8) : 3645–3650. DOI:10.1073/pnas.0909910107 |
| [13] | Xia X. Position weight matrix, Gibbs sampler, and the associated significance tests in motif characterization and prediction. Scientifica, 2012 : 917540. |
| [14] | Sharp P M, Li W H. The codon adaptation index——a measure of directional synonymous codon usage bias, and its potential applications. Nucleic Acids Res, 1987, 15(3) : 1281–1295. DOI:10.1093/nar/15.3.1281 |
| [15] | Ikemura T. Correlation between the abundance of yeast transfer RNAs and the occurrence of the respective codons in protein genes. Differences in synonymous codon choice patterns of yeast and Escherichia coli with reference to the abundance of isoaccepting transfer RNAs. J Mol Biol, 1982, 158(4) : 573–597. DOI:10.1016/0022-2836(82)90250-9 |
| [16] | Comeron J M, Aguade M. An evaluation of measures of synonymous codon usage bias. J Mol Evol, 1998, 47(3) : 268–274. DOI:10.1007/PL00006384 |
| [17] | Xia X. DAMBE5:A comprehensive software package for data analysis in molecular biology and evolution. Mol Biol Evol, 2013, 30(7) : 1720–1728. DOI:10.1093/molbev/mst064 |
| [18] | Xia X. A major controversy in codon-anticodon adaptation resolved by a new codon usage index. Genetics, 2014, 199(2) : 573–579. |
| [19] | Chithambaram S, Prabhakaran R, Xia X. Differential codon adaptation between dsDNA and ssDNA phages in Escherichia coli. Molecular Biology & Evolution, 2014, 31(6) : 1606–1617. |
| [20] | Wang M, Weiss M, Simonovic M, et al. PaxDb, a database of protein abundance averages across all three domains of life. Molecular & Cellular Proteomics, 2012, 11(8) : 492–500. |
| [21] | Wang M, Herrmann C J, Simonovic M, et al. Version 4.0 of PaxDb:Protein abundance data, integrated across model organisms, tissues, and cell-lines. Proteomics, 2015, 15(18) : 3163–3168. DOI:10.1002/pmic.201400441 |
| [22] | Schrimpf S P, Weiss M, Reiter L, et al. Comparative functional analysis of the Caenorhabditis elegans and Drosophila melanogaster proteomes. PLoS Biology, 2009, 7(3) : 616–627. |
| [23] | Baerenfaller K, Grossmann J, Grobei M A, et al. Genome-scale proteomics reveals Arabidopsis thaliana gene models and proteome dynamics. Science, 2008, 320(5878) : 938–941. DOI:10.1126/science.1157956 |
| [24] | Castellana N E, Payne S H, Shen Z, et al. Discovery and revision of Arabidopsis genes by proteogenomics. Proceedings of the National Academy of Sciences, 2008, 105(52) : 21034–21038. DOI:10.1073/pnas.0811066106 |
| [25] | Schneeberger K. Reference-guided assembly of four diverse Arabidopsis thaliana genomes. Proc Natl Acad Sci U S A, 2011, 108(25) : 10249–10254. DOI:10.1073/pnas.1107739108 |
| [26] | Theologis A, Ecker J R, Palm C J, et al. Sequence and analysis of chromosome 1 of the plant Arabidopsis thaliana. Nature, 2000, 408(6814) : 816–820. DOI:10.1038/35048500 |
| [27] | Salanoubat M, Lemcke K, Rieger M. Sequence and analysis of chromosome 3 of the plant Arabidopsis thaliana. Nature, 2000, 408(6814) : 820–822. DOI:10.1038/35048706 |
| [28] | Tabata S, Kaneko T, Nakamura Y, et al. Sequence and analysis of chromosome 5 of the plant Arabidopsis thaliana. Nature, 2000, 408(6814) : 823–826. DOI:10.1038/35048507 |
| [29] | Sato S. Complete structure of the chloroplast genome of Arabidopsis thaliana. DNA Res, 1999, 6(5) : 283–290. DOI:10.1093/dnares/6.5.283 |
| [30] | Giegé P. RNA editing in Arabidopsis mitochondria effects 441 C to U changes in ORFs. Proc Natl Acad Sci U S A, 1999, 96(26) : 15324–15329. DOI:10.1073/pnas.96.26.15324 |
| [31] | Lin X. Sequence and analysis of chromosome 2 of the plant Arabidopsis thaliana. Nature, 1999, 402(6763) : 761–768. DOI:10.1038/45471 |
| [32] | Mayer K. Sequence and analysis of chromosome 4 of the plant Arabidopsis thaliana. Nature, 1999, 402(6763) : 769–777. DOI:10.1038/47134 |
| [33] | Wright F. The effective number of codons used in a gene. Gene, 1990, 87(1) : 23–29. DOI:10.1016/0378-1119(90)90491-9 |
| [34] | Xia X. An improved implementation of codon adaptation index. Evolutionary Bioinformatics, 2007, 3(1) : 53–58. |
| [35] | Nagaraj N, Wisniewski J R, Geiger T, et al. Deep proteome and transcriptome mapping of a human cancer cell line. Mol Syst Biol, 2011, 7(1) : 548–557. |
| [36] | Hofacker I L. Vienna RNA secondary structure server. Nucleic Acids Res, 2003, 31(13) : 3429–3431. DOI:10.1093/nar/gkg599 |
| [37] | Lukow O M, Preston K R, Watts B M, et al. Measuring the influence of wheat protein in breadmaking:from damage control to genetic manipulation of protein composition in wheat. In:Wvigleyc W.Wheat Quality Elucidation. The Bushuk Legacy. St-Paul, MN:2002, 42(3):50-64. |
| [38] | Mokshina N, Gorshkova T, Deyholos M K. Chitinase-like (CTL) and cellulose synthase (CESA) gene expression in gelatinous-type cellulosic walls of flax (Linum usitatissimum L.) Bast Fibers. Plos One, 2014, 9(6) : e97949. DOI:10.1371/journal.pone.0097949 |
| [39] | Ahmed M M, Ralph Patrick, Timothy L, et al. Predicting the dynamics of protein abundance. Molecular & Cellular Proteomics, 2014, 13(5) : 1330–1340. |