 2017, Vol. 37
2017, Vol. 37文章信息
- 徐媛媛, 俞翰炳, 吴飞华, 吴晓梅.
- XU Yuan-yuan, YU Han-bing, WU Fei-hua, WU Xiao-mei.
- 基因组时代林木抗病分子机理研究的新进展
- Molecular Mechanisms of Antimicrobial Defenses and Resistance in Forest Trees in a Genomic Era
- 中国生物工程杂志, 2017, 37(6): 114-123
- China Biotechnology, 2017, 37(6): 114-123
- http://dx.doi.org/DOI:10.13523/j.cb.20170617
-
文章历史
- 收稿日期: 2016-12-08
- 修回日期: 2017-02-12






林木具有调节气候,维持生物多样性等重要的生态效益,同时也是主要的可再生生物能源物质。森林病虫害不断影响和威胁着林业的生态及经济效益。深入探究林木如何抵御病虫害及其两者间的互作机制可以加快抗病林木品种的选育,有利于进行科学防治,也可为相关部门制定林业管理政策提供数据支撑。但是,林木具有生命周期长、不易获得基因突变体等特点[1],使得其抗病分子机理的研究滞后于草本植物,尤其是拟南芥、水稻等模式植物。
木本植物的抗病分子机理与模式草本植物相似[2]。但林木作为多年生植物,其生长速度和进化速率低于一年生的草本植物,更远远低于活跃的病原菌。林木是如何利用其基因组中的有限"工具"去感知和抵御进化速度快且种类繁多的病原菌呢?这是目前林木病理学研究的重要问题之一。
近年来,高通量的基因组学和转录组学(transcriptomics)研究是林木抗病领域的热点,也是深入理解林木与微生物交互作用的强有力手段。基因组学和转录组学的应用标志着林木病理学的研究进入一个新时代。本文拟在基因组时代的背景之下,就近年来林木抗病分子机制所取得的成就,以及高通量测序技术在林木分子病理学研究中的作用与面临的挑战进行综述与分析。
1 林木抗病机制特点通过模式草本植物及其相关突变体的分子生物学研究,人们已对草本植物如何响应病原菌(包括真菌和细菌)的侵染,以及二者间的相互作用有了较为明确的认知。植物可通过病原模式识别受体(pattern recognition receptors,PRRs)来鉴别绝大多数与病原菌相关的保守分子模式(pathogen-associated molecular patterns,PAMPs),并激发植物基础免疫机制,而一些病原菌可释放效应物(effector)抑制其宿主植物的基础免疫响应。此时,植株可进一步通过抗性(resistance,R)蛋白去识别病原菌效应物及其导致的变化,产生由效应物激发的免疫[3]。这两种免疫响应通过植物激素组成的信号转导通路传递。其中,水杨酸(salicylic acid,SA)信号途径主要与植物抵抗活体营养型和半活体营养型的病原菌有关,而茉莉酸(jasmonic acid, JA)/乙烯信号途径则被认为与坏死营养型病原菌的抗性有关。这两条信号通路主要表现为拮抗作用,但也会表现出相互协同[4]。这些激素及下游转录因子将进一步调控相关基因(如编码参与合成和修饰病程相关蛋白(pathogenesis-related protein,PR)和次生代谢产物的基因)的表达,并最终决定植物的抗性水平(图 1)。

|
| 图 1 植物响应病原菌(包括细菌和真菌)侵染的示意图 Figure 1 Schematic illustration of induced responses in host cells to pathogens |
林木的抗病机制类似草本植物,但也有自身的特点。为了降低漫长生命周期中的发病率,林木增强了对病原菌的物理、化学防御。如坚韧的树皮可以保护其有活性的形成层和韧皮部,以及木质部中的木质素具有抗菌作用等。这些特性使得林木免受绝大多数病原菌的侵扰,是木本植物的第一道防线。当第一道防线被破坏时(如植食昆虫的啃咬),病原体便较容易地进入林木体内。此时,诱导性防御就会被激活[5]。木本植物感知病原菌入侵的过程类似于草本植物,也是通过受体蛋白或是R蛋白所介导[6]。不过,在信号通路下游的激素网络方面,林木与草本模式植物则表现出了较大差异。在草本植物中,SA和JA信号通路可以激发生物体的免疫响应。虽然SA和JA信号通路相关基因均在受疫霉菌(Phytophthora cinnamomi)侵染的鳄梨(Persea americana)中被诱导[7],但SA在林木中成功诱导宿主防御反应的概率并不高[8-9]。
尽管大量的生理、生化研究阐述了林木抗病的生理机制,也有一些研究通过正向遗传学和数量性状位点定位(quantitative trait loci,QTL)等传统分子生物学手段,初步探究了其背后的分子机理,但由于林木自身的特点,相关研究进展仍较为缓慢,有许多问题还需要进一步地探究,如SA信号通路在林木抗病中的作用。如今,基于高通量测序技术的多组学分析技术,大大加快了林木抗病分子机理的研究,同时也为此问题的研究提供新的视角。
2 林木的结构基因组学和抗病基因分析2006年,杨树全基因组测序的完成,标志着林木抗病分子机理的研究进入了基因组时代[10]。以全基因组测序为目标的结构基因组学有助于建立高密度的遗传、物理和转录图谱,为全面解析杨树的抗病过程提供遗传背景;功能基因组学研究则在结构基因组学提供信息的基础上进一步展开,主要内容涉及绘制基因的表达谱、鉴定基因的功能以及相关的比较基因组学研究等。杨树作为木本模式植物,对其基因组的研究可以为揭示SA信号通路在林木抗病中的作用奠定基础。美国佐治亚大学的Xue等[11]发现杨树细胞内的SA水平持续性的提升可以改变光合和氧化胁迫的状态。网络分析进一步揭示了RLK和生物/非生物胁迫的响应转录因子WRKY可能驱动SA介导的下游基因的转录。不过,拟南芥SA信号通路中的一些基因在杨树中没有体现出相应的功能。ICS基因在拟南芥生物胁迫中诱导SA合成,但在杨树中则参与合成叶绿醌。与这两种不同的抗逆策略相对应,拟南芥和杨树中的ICS基因在基因复制和可变性剪切方面也具有独立的进化模式[12]。虽然目前对杨树的基因组学研究并不能完全解释SA成功诱导林木抗病概率较低的问题,但随着功能基因组学研究方法和手段的不断更新和完善,相信在可预见的未来,这一问题会逐步得到解答。
杨树并不能代表所有的林木,尤其是亲缘关系远的裸子植物,如挪威云杉。后者的基因组大小为20 Gb,为杨树基因组的37倍[13]。这些亲缘关系不一的木本植物所敏感的病原菌并不相同,因而在感知和抵抗病原菌方面也往往存在显著的差异。这些木本植物与其病原菌的相互作用需要更深入的研究。
基因组时代下的高通量测序技术有效地解决了林木基因组大、测序价格高的问题。从2006年的杨树基因组草图完成至今,已完成全基因组测序的林木(表 1) 覆盖了裸子植物的2个属(云杉和松柏)[13-16]和被子植物的约16个属[10, 17-31]。被子植物中,只有一个单子叶林木(椰枣Phoenix dactylifera)的草图完成,其余均为双子叶林木。值得一提的是,果树类基因组占了已测序的双子叶林木的1/3,比如苹果、梨和柑橘。研究人员相信全基因组所带来的转录组学、蛋白质组和代谢组学等结构和功能分析对研究果树林木的抗病机制和提高果实产量是有帮助的。此外,很多经济林木中的常见病原菌基因组测序[32-40]也多见诸报道(表 1),这也可以为深入探讨病原菌如何抑制林木中的先天免疫机制提供研究的基础。
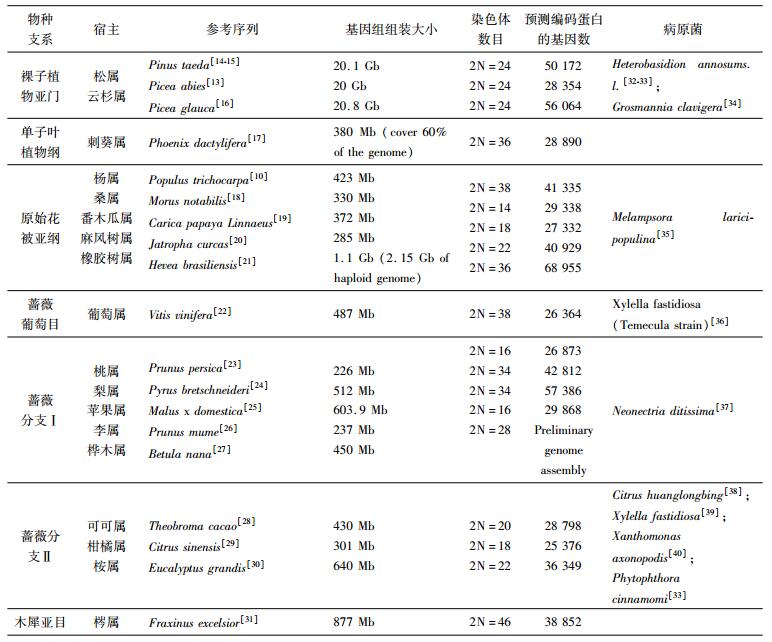
R基因是激活ETI的重要组分,其种类和数量直接决定着树种对应感病菌的种类及敏感程度。目前已克隆的绝大多数R基因是一类含有核苷酸结合位点(nucleotide-binding site,NBS)以及富含亮氨酸重复(leucine-rich repeats,LRR)的NBS-LRR基因。NBS-LRR基因编码的蛋白位于细胞质中。其中,LRR结构域可以介导受体-配体之间的相互作用,起到识别病原菌的作用;NBS结构域负责进入细胞核,激活相关基因的表达。根据NBS-LRR类基因所编码蛋白的N端结构域CC(coiled coil)或者TIR(Toll/Interleukin-1 Receptor),NBS-LRR基因又可进一步分为两种:CC-NBS-LRR(CNL)和TIR-NBS-LRR(TNL)。通过追溯进化起源,研究发现TNL类基因最早出现在苔藓类植物中,而CNL类基因起源于石松类植物中,所以TNL类基因的出现早于CNL类基因[41]。除了结构和进化起源的差异,TNL和CNL下游的信号通路也不同,使得两种NBS-LRR基因出现功能分化。
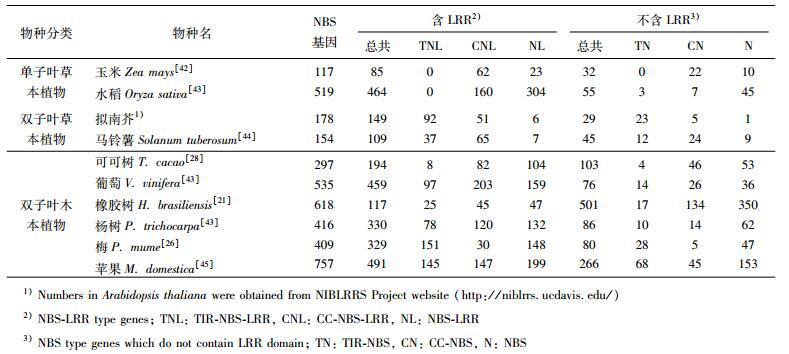
根据林木的全基因组序列信息,挖掘并预测林木的R基因家族的种类和数量,是研究林木的抗病机制的关键之一。根据目前的结构基因组学研究,我们对不同植物物种的NBS基因进行了汇总(表 2)[21, 26, 28, 42-45]。由表 2的数据可知:(1) 单子叶植物中仅发现CC-NBS-LRR基因,而大部分双子叶植物中两种NBS-LRR(TIR-NBS-LRR和CC-NBS-LRR)均存在[46]。(2) NBS基因数量的多少与基因组大小无必然联系。木本植物的R基因普遍多于草本植物的R基因,这可能为木本植物提供了更好的抗病能力。如木本植物可可树297个R基因,苹果多达757个,而草本植物拟南芥中预测的R基因是178个,玉米117个(535个水稻R基因可能与高密度的人工筛选有关)。(3) TNL蛋白N末端的TIR结构域,与果蝇Toll蛋白及哺乳动物白细胞介素受体的胞外结构域相似。有研究表明果蝇和哺乳动物的Toll和Toll同系物在动物的免疫机制中起着信号转导的作用,而植物中的TNL基因也直接与疾病的抗性有关。例如拟南芥中的TNL基因RPS4有助于植物成功抵御丁香假单胞杆菌番茄致病变种(Pseudomonas syringae pv.tomato)的侵染。TNL的起源早于裸子植物和被子植物的分歧演化,而这类基因在单子叶植物中缺失,在双子叶植物中数量也不一致。拟南芥的NBS-LRR抗性蛋白中TNL占了62%,葡萄和杨树是21%,可可树只占了4%。系统发育分析可以描述TNL基因在不同基因组中所经历的进化轨迹[28],然而关于单子叶物种中TNL基因的缺失及不同基因组中NBS类抗病基因的数量差异的具体机制,还有待进一步深入研究[47]。
|
3 林木的转录组学分析
转录组学在结构基因组学的基础上进一步研究林木抗病的分子机理,可找出受病原菌诱导的差异表达基因。目前常用的转录组学分析方法有基因芯片和转录组测序(RNA-seq)两种。基因芯片技术出现较早,在应用上也趋于成熟,使得后期数据分析十分快捷,并有完整的理论体系支撑。相对于基因芯片,RNA-seq的优势在于:(1) 无需预先知道ESTs信息,可以运用在无参考基因组序列的非模式生物中[48];(2) 产生更高的核酸分辨率,检测到表达量很低的转录物,避免了杂交所引起的问题以及允许无偏好地扫描基因组的基因(即能够检测到目标基因之外的新的潜在的差异性表达的基因),因此RNA-seq的应用潜力非常大。
基于基因芯片和第一代测序技术,林木的转录组学研究在杨树中率先展开[49-51]。就目前而言,杨树的转录组学研究结果与原先的生理生化结果基本相符。在接种后的杨树中,只有少部分病原菌相关的基因(如PR-1和几丁质酶)在整个试验周期都是上调的,而大部分基因只在一些特定的时间周期内上调。比如,在接种后期原花色素合成所需要的酶突然之间被大量合成,植物化学分析也验证了受感染的叶片中原花色素水平会提高[49]。另外,将不兼容性的叶锈病病原菌接种到杨树中,宿主的PR-1、PR-2和PR-5等基因的表达量亦显著升高[51]。
五针松是重要的单子叶林木物种。北美的五针松曾被五针松疱锈病(由松疱锈病菌Cronartium ribicola引起)全面摧毁。Liu等[52]对受其侵害的五针松品种之一西部白松(Pinus monticola)进行RNA-seq分析,de novo构建其转录组,并通过比较抗病和感病的不同西部白松叶片的转录组比较,揭示了979个显著差异性表达的基因。在抗病的幼苗中,上调基因包括一系列信号感受分子,比如编码CBL互作的激酶(CIPK)、F-box蛋白质家族、ABA受体的基因,还有与凋亡诱导因子(AIF)、FT和类枯草杆菌蛋白酶同源的基因。
苹果、梨和柑橘是重要的经济林木,病菌Erwinia amylovora侵染所引起的火疫病对全球的苹果和梨树来说是一种毁灭性灾害。最典型的症状是花、果实和叶片受火疫病菌侵害后,很快变黑褐色枯萎。苹果基因组草图和基因组注释的完成[25]使得科研人员可以利用RNA-seq技术,第一次挖掘被E. amylovora侵染的苹果花部组织细胞中的转录组变化并分析其遗传背景[53]。1 080个转录物在被病菌侵染48h之后呈现出差异性表达,包括抗病、代谢和植物生长素相关的一些基因。通过将测序短序列(reads)对应到无基因覆盖的苹果基因组区域,识别出和火疫病相关的潜在的新基因。通过RT-PCR,研究人员发现一个过氧化物酶基因家族应对E.amylovora表现低表达,推测其具有易感病的潜在功能。
果树溃疡病是由真菌Neonectria ditissima引起的另一种严重影响苹果产量的林木疾病。Ghasemkhani[37]通过对两个抗性差异较大的苹果树品种进行RNA-seq分析发现,抗性品种在注射真菌N. ditissima之后,抗病相关的基因,以及参与解毒、过氧化物酶相关的反应、苯丙醇代谢和木质化过程的基因均表现显著上调。
黄龙病是一种影响全球柑橘生产的毁灭性病害。果园里的柑橘树(Citrus sinensis L. Osb.)受到柑橘黄龙病菌(Candidatus liberibacter asiaticus)入侵之后会呈现不同症状。Martinelli等[54-55]采集这些树木的4个组织细胞(年幼和成熟的叶子和果实)进行了第二代测序。结合不同的功能分析方法(包括主成分分析、信号通路和基因富集分析及蛋白质-蛋白质相互作用网络分析),研究人员挖掘出受到疾病影响的代谢功能紊乱,以及黄龙病引起的激素串扰(hormone crosstalk)、果实-叶子信号相关联的功能失调。有意义的是,作为生物/非生物胁迫的响应转录因子WRKYs(包含WRKY70),它们在果实中的表达量高于叶子,而果实一般并不认为是受病菌侵害的首要部位。因而,该工作有利于宿主响应检测方法的改进和验证病害的短期治疗策略的制定。
白蜡树(Fraxinus excelsior)枯梢病由真菌Hymenoscyphus fraxineusyin引起,可能导致90%的欧洲大陆白蜡树种群死亡。伦敦大学玛丽皇后学院的Sollars等[31]对欧洲白蜡树进行全基因组测序,获得全长cDNA的参考序列,然后重新分析野外长期观测到的枯梢病易感的丹麦白蜡树叶片组织的转录组学测序数据[56],并对在英国和欧洲大陆的白蜡树进行全面的遗传分析,得出的主要结论是:相比欧洲大陆的白蜡树,英国的白蜡树更有可能携带抵御白蜡树枯梢病的基因。因为较低的白蜡树枯梢病易感性与较低水平的植物化学物质"环烯醚萜苷类"有关,而相比欧洲大陆的白蜡树,多数英国白蜡树生成的这类化合物较少。
另外,利用RNA-seq技术研究林木转录组学的工作还涉及柯木[57]、可可树[58]和紫薇[59]。
除了编码蛋白的基因,近年来的研究还发现Ca2+、miRNA等一些小分子物质也与林木的抗病性有关。Kanchiswamy等[60]的研究表明在接种后苹果树中,抗病品种的细胞质基质Ca2+含量高于感病品种,同时在30个Ca2+依赖性蛋白激酶(Ca2+-dependent protein kinases,CPKs)中有11个属于差异表达。另外,Chen等[61]通过对未接种和接种溃疡病的杨树cDNA文库进行Solexa测序,确定了在生物胁迫中杨树miRNA的变化情况。最后,他们发现大多数miRNA在接种后都上调,其中33个miRNA显著上调。如miR393上调了6.4倍,并且这个miRNA在冷处理下表达量也会上调。Petre等[62]利用454焦磷酸测序技术对接种后的杨树叶片进行转录组学分析,发现了一个真菌诱导的稳定高表达的硫酸盐转运体。他们认为是Melampsora larici-populina通过宿主编码机制诱导产出PtSultr3;5,这个基因通过派生出硫酸盐来改变杨树的生理状况,使宿主环境更适合病原菌的增殖。以上不同的调控分子对抗病基因的调控作用,能够保证林木基因组中保存大量具有多样性的抗病基因来抵御不断变化的病原菌的入侵,还能减少过多的抗病基因表达对基因组造成的负担。
4 林木的全基因组关联分析随着基因组学研究以及基因芯片技术的发展,全基因组关联分析(Genome Wide Association Study,GWAS)方法被应用于挖掘及鉴定大量与复杂性状相关联的遗传变异。近年来,这种方法在植物遗传学和育种相关的重要性状研究中得到了广泛的应用。关联分析是一种基于连锁不平衡来识别分子标记之间或候选基因与性状之间关系的方法。基于高通量测序技术,GWAS在林木基因组学研究中可用于挖掘与抗病性状相关的SNPs和所连锁的基因。
La Mantia等[63]利用基因分型芯片对400多个不同基因型的杨树做了GWAS。他们最终找到了与叶锈病的严重程度显著相关的40个SNPs及其关联的26个基因。其中,有一个SNP处在PRR7的第5个外显子,是一个非同义突变。在拟南芥中,PRR7属于一个生物钟转录因子的基因家族,但这个基因与该种群的物候学特征没有关联。拟南芥功能缺陷型突变体prr7在调节生物节律方面表型也不明显,双突和三突的表型也只表现出了prr5和prr9单突的表型。最新研究发现,植物生物节律也参与到先天免疫机制的调节,其中PRR7基因就是其中一个关键调控元件[64]。大多数PRR7的靶基因的表达量在黎明时达到了峰值,而此时抑制这些靶基因的PRR7蛋白含量最低,这表明PRR7通过抑制早晨表达量高的基因的转录来影响胁迫响应之类的生物性状。Iwata等[65]通过DNA分子标记获得76个日本梨Pyrus pyrifolia品种的基因型。随后的GWAS分析检测到4个标记与三种表型显著相关联。其中一个表型就是由病原菊池链格胞(Alternaria kikuchiana Tanaka)引起的梨黑斑病,与其相关联的分子标记CH04h02位于连锁群LG11,并与两个疾病易感基因Ani和Ana紧密连锁。然而水果林木中的绝大多数性状很难对应到单个基因的水平,其中一个原因是缺少所研究物种的全基因组信息[66-67]。Mariette等[66]将抗李痘病(Plum pox virus)的杏树(Prunus armeniaca)进行全基因组测序,然后将桃树(Prunus persica)基因组作为参考序列来鉴别SNP,这种基于全基因组序列的GWAS不仅验证了之前的QTL区间,还发现了一些新的位点,并且这些新发现的位点都可以对应到几个备选基因,为今后深入研究提供基础。Evans等[68]利用高通量测序建立了基因与杨树的高度、出芽率之间的关联。Kumar等[69]在苹果中利用高通量GWAS在1 200个幼苗中检测到2 500个SNPs与六个果实产量有关的表型相关联。
5 分子生物学技术在林木转基因育种中的前沿成果由于传统杂交育种过程中存在的林木育种周期长且不良性状的连锁累赘等限制,通过转基因等生物技术培育优良品种的林木的研究一直是育种和生物工程领域的热点之一[2, 70-72]。早期研究主要致力于将能够直接抑制病虫害和病原生长的几丁质酶、抗菌多肽等基因转入林木基因组中,此方法能够提高林木抗性,但是也对林木的产量和生长有一定的影响。随着植物及林木抗病分子机理研究的深入开展,研究人员把注意力转向病原识别、免疫调控等方面的基因的应用,例如将杨树中的NPR1基因过表达和将番茄中的抗病基因HcrVf2转入苹果树均获得较好的效果。此外,利用RNA干涉(RNAi)技术来成功抑制外源病毒的危害已大量应用于果树育种。比较突出的例子是抗李痘病的杏树品种(HoneySweet)已经在美国获得商业化种植的许可,该品种利用PPV的包被蛋白的反向序列形成发卡式小RNA来高效抑制病毒侵染。
在植物育种中,对特定的目的基因进行编辑,这原本是几乎不可实现的难题。近两年火爆的TALEN和CRISPR/Cas9基因编辑技术极大地扫清了定向基因编辑的障碍。目前CRISPR/Cas9技术已经应用于大量的不同真核物种的研究,实现基因组几乎任意位置基因的突变、诱导表达调控和序列插入等一系列基因编辑功能。参考文献[73]报道我国科学家高彩霞研究员的课题组成功地利用基因编辑技术同时突变多个基因拷贝和特异地突变单个基因拷贝。该课题组同时突变六倍体小麦中的MLO基因的三个拷贝,获得了对白粉病具有广谱抗性的小麦材料。日本的一个研究小组[74]也首次在苹果树中利用CRISPR/Cas9技术实现了特定基因的定向编辑。另外,通过显微操作以及基因枪等技术,已可实现DNA-free的基因组编辑方法,并且可以成功避免外源DNA片段整合到基因组中的潜在风险[75]。转基因育种中的其它一些技术难题,如关键性状的体外筛选、单倍体培育和加倍化技术等,近年来也有所突破。例如,针对欧洲白蜡树的RNA-Seq研究发现了一系列与白蜡树感染程度相关的标志基因,通过这些基因的表达量所预测的林木可能感染几率与野外的实际观测结果高度一致[31],这些标志基因或许就可以应用于育种过程中胚性组织或幼苗筛选抗病性状。另外,研究人员[76]通过失活玉米精细胞中特异表达的磷脂酶基因MTL直接诱导产生单倍体玉米,从而突破了传统花药培养的限制,据推测该技术可拓展到其他植物物种。
6 展望综上所述,林木物种的基因组学和转录组学研究提供了丰富的抗病相关信息。不过,仍然存在一些问题值得探讨。(1) RNA-seq技术和分析受到林木基因组本身的一些限制。相对于草本植物,已测序的林木基因组还是很少。在缺少高质量的林木参考基因组的情况下,林木的转录组拼接还是de novo(无参考序列),这对于一些基因组大小达20 Gb左右的松柏科林木是一个挑战。另外,林木基因组中转录物的注释还是主要依靠草本植物的基因组注释(如拟南芥、水稻或大豆)[77]。(2) 利用基因分型芯片探索林木抗病性与SNPs之间的关联性的研究并不多,依赖于高通量测序技术做的GWAS则更少。显然,利用高通量技术的GWAS研究覆盖度更广,得到的结果也更全面和精确,这对于果树的育种很有意义。(3) 转录组学和蛋白质学之间的相关性并不高,所以我们需要借助蛋白质组学技术来进一步获得表达谱的全景图并挖掘受到转录后调控的基因。而上述表达谱的变化能否改变生物内源性代谢物质的组成也还需要代谢组学的确认。
尽管如此,基因组时代林木抗病分子机理的研究还是取得了丰硕成果, 为林木分子病理学奠定了坚实的基础,在不久的将来促进传统育种和基因工程育种的发展。而近年来分子生物学领域的快速发展也使得通过转基因技术培育优良林木品种的研究如虎添翼。(1) 传统的抗病育种方法主要是分子标记辅助选择,其育种的成败很大程度上取决于分子标记的质量。以往得到的分子标记有不同方面的缺陷,利用RFLP和AFLP的方法得到的分子标记分辨率和重复率较低,利用SSR的方法得到的分子标记成本则又太高。如今,高通量测序技术能够提供大量且优质廉价的SNPs,可以大大促进传统育种的发展。另一方面,基因工程抗病育种很大程度上依赖于已报道的基因。但是,这些已报道的基因主要来自于农作物、模式植物或者微生物,并非来自于林木本身。基因组时代的研究成果则可以提供更多的林木基因组本身的抗病基因信息。(2) RNAi的引入需要转基因技术,所以出于食品及生态安全的考虑,转基因果树林木品种的大规模应用仍远远落后于科学研究[78]。虽然也有大量研究致力于开发砧木RNAi技术,这样通过韧皮部的运输,siRNA可以在接穗中发挥作用,同时避免了食用部分中转基因成分的潜在危害,但其安全性仍需要长期评估[78]。因此,同源转基因技术(cisgenesis)受到了研究人员的青睐,同源转基因就是将本物种或其近缘野生种克隆的优异等位基因导入到要改良的育种材料中。同源转基因和常规育种所要导入的目标基因的来源是相同的,因此育成后的品种同样是安全的[79],这可以最大程度地减少人们对异源基因导入的担忧。高通量测序技术的蓬勃发展使得育种学家拥有目标林木的完整的基因目录。基于高通量测序的RNA-seq和GWAS技术提升了人们对树木抗病分子机理的认识,也使得从经济林木树种或其近缘野生种克隆优质等位基因的工作变得简便容易,为同源转基因改良品种的育种研究提供大量优秀的素材。(3) 宿主林木和病原菌之间的复杂关系的理解需要评估各种性状(包括抗性)的变化和这些性状在不同环境和不同水平(单棵林木、一片森林和一个生态系统)的表达量。RNA-seq和GWAS等研究与野外的长期观测抗性数据相结合,比实验室或大棚观测到的林木抗病数据,更能全面和精确地理解林木的抗病机理。因而野外田间试验对于理解林木的遗传多样性和对病原菌的抗性和适应性依旧是非常重要的。与草本植物比较,限制林木的野外实验及其相关的林木育种的一个主要因素是林木的生长周期很长且需要大片的生长空间。这使当前林木物种的野外试验具有挑战性,Buggs的团队获得了野外长期观测丹麦白蜡树抵抗枯梢病的实验数据[56],并且利用基于全基因组序列的RNA-seq分析技术,发现环烯醚萜苷含量与白蜡树感染程度紧密相关[31],这为下一步的育种应用研究指引了全新的方向。
| [1] | Neale D B, Kremer A. Forest tree genomics:growing resources and applications. Nat Rev Genet, 2011, 12(2) : 111–122. DOI:10.1038/nrg2931 |
| [2] | Kovalchuk A, Kerio S, Oghenekaro A O, et al. Antimicrobial defenses and resistance in forest trees:challenges and perspectives in a genomic era. Annu Rev Phytopathol, 2013, 51 : 221–244. DOI:10.1146/annurev-phyto-082712-102307 |
| [3] | Jones J D, Dangl J L. The plant immune system. Nature, 2006, 444(7117) : 323–329. DOI:10.1038/nature05286 |
| [4] | Robert-Seilaniantz A, Grant M, Jones J D. Hormone crosstalk in plant disease and defense:more than just jasmonate-salicylate antagonism. Annu Rev Phytopathol, 2011, 49 : 317–343. DOI:10.1146/annurev-phyto-073009-114447 |
| [5] | Eyles A, Bonello P, Ganley R, et al. Induced resistance to pests and pathogens in trees. New Phytol, 2010, 185(4) : 893–908. DOI:10.1111/j.1469-8137.2009.03127.x |
| [6] | Duplessis S, Major I, Martin F, et al. Poplar and pathogen interactions:insights from populus genome-wide analyses of resistance and defense gene families and gene expression profiling. Crit Rev Plant Sci, 2009, 28(5) : 309–334. DOI:10.1080/07352680903241063 |
| [7] | Reeksting B J, Coetzer N, Mahomed W, et al. De novo sequencing, assembly, and analysis of the root transcriptome of Persea americana (Mill.) in response to Phytophthora cinnamomi and flooding. PLoS One, 2014, 9(2) : e86399. DOI:10.1371/journal.pone.0086399 |
| [8] | Arnerup J, Lind M, Olson A, et al. The pathogenic white-rot fungus Heterobasidion parviporum triggers non-specific defence responses in the bark of Norway spruce. Tree Physiol, 2011, 31(11) : 1262–1272. DOI:10.1093/treephys/tpr113 |
| [9] | Arnerup J, Nemesio-Gorriz M, Lunden K, et al. The primary module in Norway spruce defence signalling against H. annosum s.l. seems to be jasmonate-mediated signalling without antagonism of salicylate-mediated signalling. Planta, 2013, 237(4) : 1037–1045. DOI:10.1007/s00425-012-1822-8 |
| [10] | Tuskan G A, DiFazio S, Jansson S, et al. The genome of black cottonwood, Populus trichocarpa (Torr. & Gray). Science, 2006, 313(5793) : 1596–1604. DOI:10.1126/science.1128691 |
| [11] | Xue L J, Guo W, Yuan Y, et al. Constitutively elevated salicylic acid levels alter photosynthesis and oxidative state but not growth in transgenic populus. Plant Cell, 2013, 25(7) : 2714–2730. DOI:10.1105/tpc.113.112839 |
| [12] | Yuan Y, Chung J D, Fu X, et al. Alternative splicing and gene duplication differentially shaped the regulation of isochorismate synthase in Populus and Arabidopsis. Proc Natl Acad Sci USA, 2009, 106(51) : 22020–22025. DOI:10.1073/pnas.0906869106 |
| [13] | Nystedt B, Street N R, Wetterbom A, et al. The Norway spruce genome sequence and conifer genome evolution. Nature, 2013, 497(7451) : 579–584. DOI:10.1038/nature12211 |
| [14] | Zimin A, Stevens K A, Crepeau M, et al. Sequencing and assembly of the 22-Gb loblolly pine genome. Genetics, 2014, 196(3) : 875–890. DOI:10.1534/genetics.113.159715 |
| [15] | Wegrzyn J L, Liechty J D, Stevens K A, et al. Unique features of the loblolly pine (Pinus taeda L.) megagenome revealed through sequence annotation. Genetics, 2014, 196(3) : 891–909. DOI:10.1534/genetics.113.159996 |
| [16] | Birol I, Raymond A, Jackman S D, et al. Assembling the 20 Gb white spruce (Picea glauca) genome from whole-genome shotgun sequencing data. Bioinformatics, 2013, 29(12) : 1492–1497. DOI:10.1093/bioinformatics/btt178 |
| [17] | Al-Dous E K, George B, Al-Mahmoud M E, et al. De novo genome sequencing and comparative genomics of date palm (Phoenix dactylifera). Nat Biotechnol, 2011, 29(6) : 521–527. DOI:10.1038/nbt.1860 |
| [18] | He N, Zhang C, Qi X, et al. Draft genome sequence of the mulberry tree Morus notabilis. Nat Commun, 2013, 4 : 2445. |
| [19] | Ming R, Hou S, Feng Y, et al. The draft genome of the transgenic tropical fruit tree papaya (Carica papaya Linnaeus). Nature, 2008, 452(7190) : 991–996. DOI:10.1038/nature06856 |
| [20] | Sato S, Hirakawa H, Isobe S, et al. Sequence analysis of the genome of an oil-bearing tree, Jatropha curcas L. DNA Res, 2011, 18(1) : 65–76. DOI:10.1093/dnares/dsq030 |
| [21] | Rahman A Y, Usharraj A O, Misra B B, et al. Draft genome sequence of the rubber tree Hevea brasiliensis. BMC Genomics, 2013, 14 : 75. DOI:10.1186/1471-2164-14-75 |
| [22] | Velasco R, Zharkikh A, Troggio M, et al. A high quality draft consensus sequence of the genome of a heterozygous grapevine variety. PLoS One, 2007, 2(12) : e1326. DOI:10.1371/journal.pone.0001326 |
| [23] | Verde I, Abbott A G, Scalabrin S, et al. The high-quality draft genome of peach (Prunus persica) identifies unique patterns of genetic diversity, domestication and genome evolution. Nat Genet, 2013, 45(5) : 487–494. DOI:10.1038/ng.2586 |
| [24] | Wu J, Wang Z, Shi Z, et al. The genome of the pear (Pyrus bretschneideri Rehd.). Genome Res, 2013, 23(2) : 396–408. DOI:10.1101/gr.144311.112 |
| [25] | Velasco R, Zharkikh A, Affourtit J, et al. The genome of the domesticated apple (Malus×domestica Borkh.). Nat Genet, 2010, 42(10) : 833–839. DOI:10.1038/ng.654 |
| [26] | Zhang Q, Chen W, Sun L, et al. The genome of Prunus mume. Nat Commun, 2012, 3 : 1318. DOI:10.1038/ncomms2290 |
| [27] | Wang N, Thomson M, Bodles W J, et al. Genome sequence of dwarf birch (Betula nana) and cross-species RAD markers. Mol Ecol, 2013, 22(11) : 3098–3111. DOI:10.1111/mec.12131 |
| [28] | Argout X, Salse J, Aury J M, et al. The genome of Theobroma cacao. Nat Genet, 2011, 43(2) : 101–108. DOI:10.1038/ng.736 |
| [29] | Xu Q, Chen L L, Ruan X A, et al. The draft genome of sweet orange (Citrus sinensis). Nat Genet, 2013, 45(1) : 59–66. |
| [30] | Myburg A A, Grattapaglia D, Tuskan G A, et al. The genome of Eucalyptus grandis. Nature, 2014, 510(7505) : 356–362. |
| [31] | Sollars E S, Harper A L, Kelly L J, et al. Genome sequence and genetic diversity of European ash trees. Nature, 2017, 541(7636) : 212–216. |
| [32] | Olson A, Aerts A, Asiegbu F, et al. Insight into trade-off between wood decay and parasitism from the genome of a fungal forest pathogen. New Phytol, 2012, 194(4) : 1001–1013. DOI:10.1111/nph.2012.194.issue-4 |
| [33] | Nordberg H, Cantor M, Dusheyko S, et al. The genome portal of the Department of Energy Joint Genome Institute:2014 updates. Nucleic Acids Res, 2014, 42(Database issue) : D26–31. |
| [34] | DiGuistini S, Wang Y, Liao N Y, et al. Genome and transcriptome analyses of the mountain pine beetle-fungal symbiont Grosmannia clavigera, a lodgepole pine pathogen. Proc Natl Acad Sci USA, 2011, 108(6) : 2504–2509. DOI:10.1073/pnas.1011289108 |
| [35] | Duplessis S, Cuomo C A, Lin Y C, et al. Obligate biotrophy features unraveled by the genomic analysis of rust fungi. Proc Natl Acad Sci USA, 2011, 108(22) : 9166–9171. DOI:10.1073/pnas.1019315108 |
| [36] | Van Sluys M A, de Oliveira M C, Monteiro-Vitorello C B, et al. Comparative analyses of the complete genome sequences of Pierce's disease and citrus variegated chlorosis strains of Xylella fastidiosa. J Bacteriol, 2003, 185(3) : 1018–1026. DOI:10.1128/JB.185.3.1018-1026.2003 |
| [37] | Ghasemkhani M. Resistance against fruit tree canker in apple-evaluation of disease symptoms, histopathological and RNA-Seq analyses in different cultivars, genetic variation of Neonectria ditissima. Alnarp:Swedish University of Agricultural Sciences, 2015, 77 : 1–64. |
| [38] | Duan Y P, Zhou L J, Hall D G, et al. Complete genome sequence of citrus huanglongbing bacterium, 'Candidatus Liberibacter asiaticus' obtained through metagenomics. Mol Plant Microbe Interact, 2009, 22(8) : 1011–1020. DOI:10.1094/MPMI-22-8-1011 |
| [39] | Simpson A J, Reinach F C, Arruda P, et al. The genome sequence of the plant pathogen Xylella fastidiosa. Nature, 2000, 406(6792) : 151–159. DOI:10.1038/35018003 |
| [40] | da Silva A C, Ferro J A, Reinach F C, et al. Comparison of the genomes of two Xanthomonas pathogens with differing host specificities. Nature, 2002, 417(6887) : 459–463. DOI:10.1038/417459a |
| [41] | Yue J X, Meyers B C, Chen J Q, et al. Tracing the origin and evolutionary history of plant nucleotide-binding site-leucine-rich repeat (NBS-LRR) genes. New Phytol, 2012, 193(4) : 1049–1063. DOI:10.1111/j.1469-8137.2011.04006.x |
| [42] | Li J, Ding J, Zhang W, et al. Unique evolutionary pattern of numbers of gramineous NBS-LRR genes. Mol Genet Genomics, 2010, 283(5) : 427–438. DOI:10.1007/s00438-010-0527-6 |
| [43] | Yang S, Zhang X, Yue J X, et al. Recent duplications dominate NBS-encoding gene expansion in two woody species. Mol Genet Genomics, 2008, 280(3) : 187–198. DOI:10.1007/s00438-008-0355-0 |
| [44] | Lozano R, Ponce O, Ramirez M, et al. Genome-wide identification and mapping of NBS-encoding resistance genes in Solanum tuberosum group phureja. PLoS One, 2012, 7(4) : e34775. DOI:10.1371/journal.pone.0034775 |
| [45] | Arya P, Kumar G, Acharya V, et al. Genome-wide identification and expression analysis of NBS-encoding genes in Malus×domestica and expansion of NBS genes family in Rosaceae. PLoS One, 2014, 9(9) : e107987. DOI:10.1371/journal.pone.0107987 |
| [46] | Bai J, Pennill L A, Ning J, et al. Diversity in nucleotide binding site-leucine-rich repeat genes in cereals. Genome Res, 2002, 12(12) : 1871–1884. DOI:10.1101/gr.454902 |
| [47] |
罗莎.
植物NBS类抗病基因的进化. 遗传, 2014, 36(12) : 1219–1225.
Luo S. Evolution of plant NBS encoding disease resistance genes. Hereditas, 2014, 36(12) : 1219–1225. |
| [48] | Gongora-Castillo E, Buell C R. Bioinformatics challenges in de novo transcriptome assembly using short read sequences in the absence of a reference genome sequence. Nat Prod Rep, 2013, 30(4) : 490–500. DOI:10.1039/c3np20099j |
| [49] | Miranda M, Ralph S G, Mellway R, et al. The transcriptional response of hybrid poplar (Populus trichocarpa x P. deltoides) to infection koby Melampsora medusae leaf rust involves induction of flavonoid pathway genes leading to the accumulation of proanthocyanidins. Mol Plant Microbe Interact, 2007, 20(7) : 816–831. DOI:10.1094/MPMI-20-7-0816 |
| [50] | Azaiez A, Boyle B, Levee V, et al. Transcriptome profiling in hybrid poplar following interactions with melampsora rust fungi. Mol Plant Microbe Interact, 2009, 22(2) : 190–200. DOI:10.1094/MPMI-22-2-0190 |
| [51] | Rinaldi C, Kohler A, Frey P, et al. Transcript profiling of poplar leaves upon infection with compatible and incompatible strains of the foliar rust Melampsora larici-populina. Plant Physiol, 2007, 144(1) : 347–366. DOI:10.1104/pp.106.094987 |
| [52] | Liu J J, Sturrock R N, Benton R. Transcriptome analysis of Pinus monticola primary needles by RNA-seq provides novel insight into host resistance to Cronartium ribicola. BMC Genomics, 2013, 14 : 884. DOI:10.1186/1471-2164-14-884 |
| [53] | Kamber T, Buchmann J P, Pothier J F, et al. Fire blight disease reactome:RNA-seq transcriptional profile of apple host plant defense responses to Erwinia amylovora pathogen infection. Sci Rep, 2016, 6 : 21600. DOI:10.1038/srep21600 |
| [54] | Martinelli F, Reagan R L, Uratsu S L, et al. Gene regulatory networks elucidating huanglongbing disease mechanisms. PLoS One, 2013, 8(9) : e74256. DOI:10.1371/journal.pone.0074256 |
| [55] | Martinelli F, Uratsu S L, Albrecht U, et al. Transcriptome profiling of citrus fruit response to huanglongbing disease. PLoS One, 2012, 7(5) : e38039. DOI:10.1371/journal.pone.0038039 |
| [56] | Harper A L, McKinney L V, Nielsen L R, et al. Molecular markers for tolerance of European ash (Fraxinus excelsior) to dieback disease identified using Associative Transcriptomics. Sci Rep, 2016, 6 : 19335. DOI:10.1038/srep19335 |
| [57] | Hayden K J, Garbelotto M, Knaus B J, et al. Dual RNA-seq of the plant pathogen Phytophthora ramorum and its tanoak host. Tree Genet Genom, 2014, 10(3) : 489–502. DOI:10.1007/s11295-014-0698-0 |
| [58] | Teixeira P J, Thomazella D P, Reis O, et al. High-resolution transcript profiling of the atypical biotrophic interaction between Theobroma cacao and the fungal pathogen Moniliophthora perniciosa. Plant Cell, 2014, 26(11) : 4245–4269. DOI:10.1105/tpc.114.130807 |
| [59] | Wang X, Shi W, Rinehart T. Transcriptomes that confer to plant defense against powdery mildew disease in Lagerstroemia indica. Int J Genomics, 2015, 2015 : 528395. |
| [60] | Kanchiswamy C N, Mohanta T K, Capuzzo A, et al. Differential expression of CPKs and cytosolic Ca2+ variation in resistant and susceptible apple cultivars (Malus x domestica) in response to the pathogen Erwinia amylovora and mechanical wounding. BMC Genomics, 2013, 14 : 760. DOI:10.1186/1471-2164-14-760 |
| [61] | Chen L, Ren Y Y, Zhang Y Y, et al. Genome-wide profiling of novel and conserved Populus microRNAs involved in pathogen stress response by deep sequencing. Planta, 2012, 235(5) : 873–883. DOI:10.1007/s00425-011-1548-z |
| [62] | Petre B, Morin E, Tisserant E, et al. RNA-seq of early-infected poplar leaves by the rust pathogen Melampsora larici-populina uncovers PtSultr3;5, a fungal-induced host sulfate transporter. PLoS One, 2012, 7(8) : e44408. DOI:10.1371/journal.pone.0044408 |
| [63] | La Mantia J, Klapste J, El-Kassaby Y A, et al. Association analysis identifies Melampsora xcolumbiana poplar leaf rust resistance SNPs. PLoS One, 2013, 8(11) : e78423. DOI:10.1371/journal.pone.0078423 |
| [64] | Zhou M, Wang W, Karapetyan S, et al. Redox rhythm reinforces the circadian clock to gate immune response. Nature, 2015, 523(7561) : 472–476. DOI:10.1038/nature14449 |
| [65] | Iwata H, Hayashi T, Terakami S, et al. Potential assessment of genome-wide association study and genomic selection in Japanese pear Pyrus pyrifolia. Breed Sci, 2013, 63(1) : 125–140. DOI:10.1270/jsbbs.63.125 |
| [66] | Mariette S, Wong Jun Tai F, Roch G, et al. Genome-wide association links candidate genes to resistance to Plum Pox Virus in apricot (Prunus armeniaca). New Phytol, 2016, 209(2) : 773–784. DOI:10.1111/nph.13627 |
| [67] | Iwata H, Minamikawa M F, Kajiya-Kanegae H, et al. Genomics-assisted breeding in fruit trees. Breed Sci, 2016, 66(1) : 100–115. DOI:10.1270/jsbbs.66.100 |
| [68] | Evans L M, Slavov G T, Rodgers-Melnick E, et al. Population genomics of Populus trichocarpa identifies signatures of selection and adaptive trait associations. Nat Genet, 2014, 46(10) : 1089–1096. DOI:10.1038/ng.3075 |
| [69] | Kumar S, Garrick D J, Bink M C, et al. Novel genomic approaches unravel genetic architecture of complex traits in apple. BMC Genomics, 2013, 14 : 393. DOI:10.1186/1471-2164-14-393 |
| [70] | Rai M K, Shekhawat N S. Recent advances in genetic engineering for improvement of fruit crops. Plant Cell Tiss Org Cult, 2014, 116(1) : 1–15. DOI:10.1007/s11240-013-0389-9 |
| [71] | Gambino G, Gribaudo I. Genetic transformation of fruit trees:current status and remaining challenges. Transgenic Res, 2012, 21(6) : 1163–1181. DOI:10.1007/s11248-012-9602-6 |
| [72] |
Powell W A, Maynard C A, Boyle B, et al. Fungal and Bacterial Resistance in Transgenic Trees. In:Tree Transgenesis. Fladung M, Ewald D, ed. Tree Transgenesis. Berlin/Heidelberg:Springer, 2006. 235-252. |
| [73] | Wang Y, Cheng X, Shan Q, et al. Simultaneous editing of three homoeoalleles in hexaploid bread wheat confers heritable resistance to powdery mildew. Nat Biotechnol, 2014, 32(9) : 947–951. DOI:10.1038/nbt.2969 |
| [74] | Nishitani C, Hirai N, Komori S, et al. Efficient genome editing in apple using a CRISPR/Cas9 system. Sci Rep, 2016, 6 : 31481. DOI:10.1038/srep31481 |
| [75] | Zhang Y, Liang Z, Zong Y, et al. Efficient and transgene-free genome editing in wheat through transient expression of CRISPR/Cas9 DNA or RNA. Nature Communications, 2016, 7 : 12617. DOI:10.1038/ncomms12617 |
| [76] | Kelliher T, Starr D, Richbourg L, et al. Matrilineal, a sperm-specific phospholipase, triggers maize haploid induction. Nature, 2017, 542(7639) : 105–109. DOI:10.1038/nature20827 |
| [77] | Heredia U L, Vazquez-Poletti J L. RNA-seq analysis in forest tree species:bioinformatic problems and solutions. Tree Genet Genom, 2016, 12(2) : 30. DOI:10.1007/s11295-016-0995-x |
| [78] | Lemgo G N, Sabbadini S, Pandolfini T, et al. Biosafety considerations of RNAi-mediated virus resistance in fruit-tree cultivars and in rootstock. Transgenic Res, 2013, 22(6) : 1073–1088. DOI:10.1007/s11248-013-9728-1 |
| [79] |
黄三文, 杜永臣, 屈冬玉, 等.
同源转基因将成为利用野生资源进行作物育种的一种有效手段. 园艺学报, 2006, 33(6) : 1397–1400.
Huang S W, Du Y C, Qu D Y, et al. Cisgenesis will become an effective tool of crop improvement in use of wild germplasm. Acta Hortic Sin, 2006, 33(6) : 1397–1400. |