 2017, Vol. 37
2017, Vol. 37文章信息
- 陈蓉, 杨帆, 成细瑶, 苏正定.
- CHEN Rong, YANG Fan, CHENG Xi-yao, SU Zheng-ding.
- 模拟α-螺旋多肽类似物抑制剂设计的研究进展
- Progress in Designing of Inhibitors Mimicking α-Helical Peptide
- 中国生物工程杂志, 2017, 37(4): 89-97
- China Biotechnology, 2017, 37(4): 89-97
- http://dx.doi.org/DOI:10.13523/j.cb.20170412
-
文章历史
- 收稿日期: 2016-08-03
- 修回日期: 2017-02-07






α-螺旋是最常见的蛋白质二级结构单元之一,其结构特征为肽链的主链围绕一个虚拟轴以螺旋的形式延伸。分析蛋白质结构数据库 (PDB) 中蛋白-蛋白复合物的结构发现,多达62%的蛋白质-蛋白质相互作用 (PPI) 界面以α-螺旋结构为主体[1]。通过丙氨酸扫描突变实验分析α-螺旋上的关键氨基酸残基,发现α-螺旋上仅需要少数热点氨基酸残基 (hotspots) 与伴侣蛋白质 (partner) 表面的疏水口袋相互作用,形成蛋白质复合物,来调控相应的功能[2]。传统上,为获得亲和力高的抑制剂,通常通过反复、高通量地筛选化合物库。这种方法存在着一定的机遇性。通过模拟α-螺旋骨架及其hotspots官能团,进行理性设计小分子抑制剂,获得抑制PPI的先导药物化合物,达到干扰蛋白质相互作用的目的。
模拟α-螺旋设计小分子抑制剂,主要采取两种策略:第一,可以将生物活性肽经过结构优化获得模拟α-螺旋的肽类或拟肽类化合物;第二,针对小分子化合物分子量小、生物稳定性高的优势,也可以通过小分子化合物来模拟。
1 肽类模拟α-螺旋骨架结构噬菌体展示技术是筛选合适多肽类骨架的快速方法。在筛选p53-Mdm2抑制剂时,首先通过噬菌体展示技术筛选得到先导肽类骨架 (Ac-Gln-Glu-Thr-Phe19-Ser-Asp-Leu-Trp23-Lys-Leu-Leu26-Pro-NH2),其活性较差 (IC50为8.7μM),通过一系列α-螺旋结构模拟优化,得到活性较强 (IC50为5nM) 的多肽类似物[Ac-Phe19-Met-Asp-Pmp-(6-Cl) Trp23-Glu-Ac3c-Leu26-NH2][3],这些类似物模拟了p53多肽的3个关键氨基酸残基 (hotspots) Phe19、Trp23、Leu26,这表明肽类类似物在对hotspots的模拟上具有明显的优势。
通过肽类化合物模拟α-螺旋设计多肽类抑制剂简单直观,且多肽类似物能够高度保留复杂的功能基团。但多肽化合物本身作为药物存在部分问题,如药代动力学性质差,容易被体内的蛋白水解酶降解等,且人工合成的多肽与母体肽段相比在水溶液中呈随机松散构象,这严重阻碍了多肽类似物与靶蛋白之间的相互作用。综上所述,在模拟α-螺旋设计肽类化合物时,必须要解决如何保持短肽的螺旋性和降低蛋白水解酶的敏感度等问题[4]。
1.1 肽类化合物的螺旋性为了保持短肽在水溶液中的螺旋性,已经研发出多种可行方法,可以通过氢键、二硫键、内酰胺键、多肽N-端修饰等多种方法对短肽进行固定。以下主要概述几种典型的稳定多肽螺旋的方法。
1.1.1 氢键替代氢键在维持α-螺旋稳定上起关键作用,通过α-螺旋上主链间的相互作用稳定螺旋,α-螺旋具有重复的 (i,i+4) 氢键[5]。为了维持肽类化合物的螺旋性,会通过共价键代替氢键,更强地固定肽链上的螺旋,稳定螺旋的结构,且是在螺旋内部进行修饰,对多肽与靶蛋白相互作用影响较小。
2005年,研究者通过人工合成的氢键 (HBS) 代替氢键固定螺旋,发现通过HBS这种共价结合的方式能更好地稳定螺旋,且这种方法对多种类型的模拟蛋白质设计的多肽抑制剂都适用[6-7]。
1.1.2 二硫键Pellegrini等[8]首次提出通过二硫键来固定螺旋的方法,通过两条存在两个半胱氨酸的十二个氨基酸 (Ac-Ala-Val-Ser-Glu-Cys-Gln-Leu-Cys-His-Asp-Lys-Gly-NH2) 的多肽证明,二硫键能够固定多肽与多肽所处的环境相关,即说明是可以通过二硫键来固定螺旋。Shiau等[9]解析出雌激素受体和短肽片段 (从核受体共激活因子衍生) 的晶体结构并说明这个短肽片段形成了一个两转角两亲性的螺旋结构与雌激素受体相互作用 (图 1a)。为了模拟这种稳定的螺旋构象,Galande等[10]提出可以通过二硫键固定一个短肽片段 (LXXLL) 使其形成一个稳定的两亲性 (即亲水性和疏水性) 螺旋结构,与雌激素受体配体结合区域上的疏水性凹槽结合 (Ki=70pM)。这一结果表明通过二硫键来固定肽段确实能够维持合成的肽段以α-螺旋构象与靶蛋白相结合。
1.1.3 内酰胺键固定Sia等[11]发现,用内酰胺键交联位于i和i+7位置的两个谷氨酸残基,能够稳定长度为14个氨基酸的C肽与HIV-1gp41疏水口袋的相互作用 (图 1b),晶体结构也证实它几乎与天然的C肽保持着一样的螺旋结构。

|
| 图 1 模拟α-螺旋设计的肽类抑制剂与蛋白的复合物结构模型 Figure 1 A model of protein in complex with peptide inhibitor mimicking α-helical structure (a) Short peptide fragments:magenta, PDB ID:3ERD (b) Lactam bridge: blue, PDB ID:1GZL (c)α/β-peptide foldmer Light blue: α-amino acid; deep blue: β-amino acid, PDB ID:3FDM |
近几年,通过对多肽片段的N-端或C-端进行修饰来维持α-螺旋的结构,已经成为PPI抑制剂研究的一个热点。
Mauran等[12]通过将脂肪族N, N′-尿素低聚物连接在多肽片段的N-端或C-端或两端将其作为成核模板,使多肽片段形成为一个可以预测的并且结构稳定的螺旋结构,类似于α-螺旋,这种方法在模拟α-螺旋设计蛋白质与蛋白质相互作用的抑制剂方面是非常有潜力的一种策略。
在α-螺旋的N-端经常出现脯氨酸,脯氨酸决定α-螺旋上第一个羰基的朝向,它也促进螺旋的形成过程,脯氨酸在α-螺旋的形成过程中起重要作用。2016年,Li等[13](李子刚课题组) 通过将合成得到的顺式-4-氨基-L-脯氨酸衍生物引入到多肽的N-末端,该衍生物可以通过共价键固相内环化,这种反应模式促进了螺旋多肽的形成。将顺式-4-氨基-L-脯氨酸衍生物引入N-端,利用脯氨酸的特性,也通过內环化形成共价键替代氢键这两种策略协同作用稳定多肽α-螺旋的构象。
1.2 蛋白水解酶的影响α-氨基酸是天然蛋白质的主要组成成分,而除了α-氨基酸之外,另外还存在β-氨基酸和γ-氨基酸,α、β、γ-氨基酸的骨架空间大小是依次增长的。α-氨基酸因为结构的特殊性,容易被蛋白水解酶降解,于是考虑用合成的β-氨基酸替代α-氨基酸,以便更好地模拟α-螺旋结构的同时,降低对蛋白水解酶的敏感性,避免多肽被降解[14]。而大量β-多肽 (β-氨基酸缩合形成的多肽) 结构和合成方法的经验总结对利用β-多肽模拟α-螺旋的研究打下了足够的基础[15]。
文献[16]报道,2009年,Gellman研究组公布了一个能够模拟Bim-BH3肽段的α-螺旋与抗凋亡蛋白Bcl-XL作用的α/β-肽段折叠体 (图 1c) 复合物的晶体结构。它们的晶体结构表明,这个折叠体有效地模拟了BH3天然的α-螺旋结构,这说明这种将天然的α-螺旋的特定位置替换为β-氨基酸的方法,不仅能够有效模拟α-螺旋的构象,而且能够明显提高与伴侣蛋白的亲和力 (Ki=2nM),另外,与Bim-BH3肽段相比,折叠体对蛋白水解酶的敏感性也明显有所降低。
考虑到肽类抑制剂细胞透膜能力弱,在体内容易被蛋白水解酶降解这一因素,Liu等[17]合成得到α-DPMI (Ac-Thr-Asn-Trp-Tyr-Ala-Asn-Leu-Glu-Lys-Leu-Leu-Arg-NH2) 和γ-DPMI (Ac-Asp-Trp-Trp-Pro-Leu-Ala-Phe-Glu-Ala-Leu-Leu-Arg-NH2) 两个十二肽化合物,尝试用脂质体包裹α-DPMI进行肿瘤生长抑制实验,MTT比色法测得其在人胶质瘤U87(WT p53) 细胞株中的IC50为1.9μM,不用脂质体包裹时其IC50为100μM,即说明可以通过脂质体包裹降低对蛋白水解酶的影响。另外实验也测得γ-DPMI不易被蛋白水解酶降解, 这进一步说明了用β或γ-氨基酸代替α-氨基酸可以降低多肽对蛋白水解酶的敏感性。
2016年,Grison等[18]以反式-2-氨基环丁基甲酸为关键的β-氨基酸组份设计合成了α/β/γ-多肽折叠体结构来模拟天然的α-螺旋,这种合成的α/β/γ-折叠体结构和野生型的α-多肽相比明显提高了对蛋白水解酶的稳定性,并且能够抑制Mdm2的活性。
利用肽类化合物来模拟α-螺旋作为抑制剂来干扰PPI已经取得了一定的成果,但是这种方法也存在很多局限,例如,由于多肽骨架的特异性,它是针对某一种靶蛋白所筛选的抑制剂骨架结构,只能顺应这一种靶蛋白的结合面,不具有普适性。而利用小分子来模拟α-螺旋作为PPI抑制剂存在着很多天然的优势。
2 小分子化合物模拟α-螺旋利用小分子模拟α-螺旋需要对螺旋骨架和hotspots两个部分进行模拟。虽然在对螺旋骨架的模拟上存在着一定的难度,但是因为小分子易于合成,分子量小,膜透过率高,具有更好的体内稳定性和成药性等优势,利用小分子模拟α-螺旋又势在必行。通过小分子来模拟α-螺旋骨架,并在小分子特定的位点连上取代基,作为α-螺旋的侧链,这是利用小分子模拟α-螺旋的核心思想。在此基础上,建立了一种模块式方法,即首先合成一个能够模拟α-螺旋的骨架,在此骨架的基础上,根据hotspots组合20种氨基酸残基存在的可能排列方式,建立模型分子库,来靶向特定靶标。
α-螺旋是通过肽链主链上第i位氨基酸残基上的-C=O与i+4位残基上的-NH之间形成氢键来稳定的,螺旋每隔3.4个残基上升一圈,高度增加0.54nm,每个残基i与相隔3(i+3) 或4(i+4) 的残基处于相似的方位 (图 2a, b, c),所以α-螺旋的侧链残基可以认为分布在三个不同的面[3]。有研究小组通过丙氨酸突变扫描 (Alanine scanning mutagenesis) 方法分析统计过hotspots在α螺旋上的分布位置,结果表明,在统计的480个以α-螺旋为对象的相互作用模型中,约60%的hotspots位于α-螺旋一面 (图 2d),约1/3的位于螺旋的两面 (图 2e),所有三面上都有的少于10%[1](图 2f)。而能否找到合适的小分子模拟α-螺旋骨架就在于能否恰好在对应的三个面连上合适的侧链来模拟hotspots。

|
| 图 2 α-螺旋上的氨基酸残基 (a) 及侧链朝向分布图 (b、c);α-螺旋上hotspots的一面 (PDB ID:1XL3)(d)、两面 (PDB ID:1XIU)(e) 和三面 (PDB ID: 1OR7)(f) 分布的案例 Figure 2 Distribution of amino acid residues on α-helix (a) and orientation of side chains (b, c);examples of the distribution of hotspots on one face (PDB ID:1XL3) (d), two faces (PDB ID:1XIU) (e) and three face in helix (PDB ID: 1OR7) (f) |
许多研究组在合成小分子来模拟α-螺旋研究方面进行了不懈的尝试,以下是几种典型的小分子模拟α-螺旋的骨架结构及其hotspots的空间分布案例 (表 1)。
| 类型 | 骨架结构类别 | 结构式 | hotspots位点 | 主要特征 | 备注 |
| hotspots一面模拟 | 三联苯类 | 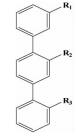 | i; i+3;i+7 | 结构简单,第一个模拟α-螺旋骨架设计的小分子 | 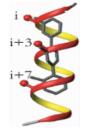 |
| 苯烯胺酮类 | 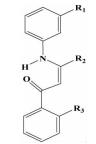 | i; i+4;i+7 | 可以对化合物中基团进行进一步的衍生,靶向其他结合位点 | ||
| 联苯N, N-二甲基甲酰胺 | 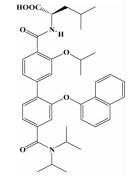 | i; i+4;i+7;i+11 | 在之前模拟螺旋两圈 (i,i+3,i+4,i+7位残基) 的基础上,实现了对螺旋第三圈 (i+11位残基) 的模拟,能靶向更多结合位点 | 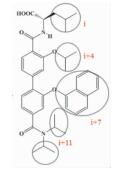 | |
| 三联吡啶类 | 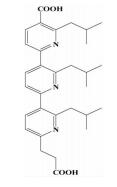 | i; i+3或i+4;i+7 | 拓宽了螺旋模拟的思路,取代苯环用芳香基衍生物作为螺旋骨架 | 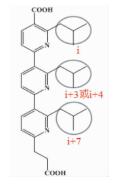 | |
| 三吡啶酰胺类 | 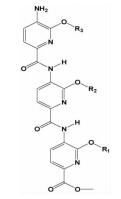 | i; i+4;i+7 | 完善了吡啶类骨架,亲水性更好,相比三联苯类化合物更易合成 | ||
| 阶梯状多环醚类 | 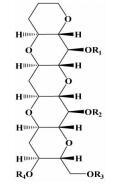 | i; i+4;i+8 | 一类新的化合物模拟骨架,能模拟α-螺旋的不同侧链 | ||
| 咪唑类 |  | i; i+3;i+7 | 这种三取代的咪唑骨架合成简单,可以在任意侧链连上取代基,有明显的潜在优势 | ||
| hotspots两面模拟 | 二联苯类 | 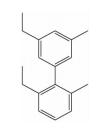 | i; i+1;i+3;i+4 | 实现了对螺旋两面上热点残基的模拟, | 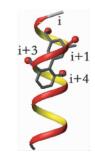 |
| 联苯茚类 |  | i; i+3;i+4;i+7 | 在三联苯的基础上,增加了一个取代位点,恰好能模拟螺旋的i+4位残基 | ||
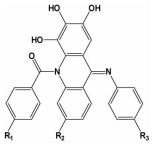 | i; i+3;i+5;i+7 | 能模拟α-螺旋的三面两圈构象,选择性和特异性是更优的 |  | ||
| hotspots三面模拟 | 十字型骨架 | 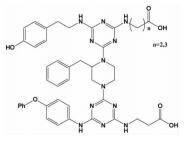 | i-2;i; i+3;i+5;i+7 | 在已有小分子骨架的基础上,在相反一面介导引入合适的带电残基,实现两亲性的三面模拟 | 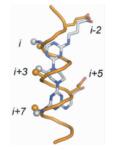 |
2.1 模拟hotspots位于α-螺旋一面的小分子骨架 2.1.1 三联苯类
Orner等[19]在对比α-螺旋上侧链与3, 2′, 2″三联苯衍生物三个功能基团取代基的俯视图时发现它们之间分布相对位置是相似的,而且当三联苯上的中间苯环与另两个苯环的二面角分别为68°和36°时,其与α-螺旋上i,i+3和i+7三个氨基酸残基的朝向高度匹配,重要的是三联苯的结构相对简单,Orner等人以此三联苯为骨架对α-螺旋进行了一系列的模拟。
该研究组[20]以此三联苯为骨架来设计小分子化合物,抑制p53-MdmX在体内的结合从而恢复p53在体内的活性。用酶联免疫吸附实验 (ELISA) 分析得到合成的小分子化合物IC50值在10~20μM之间,并且,在细胞培养中当小分子浓度达到15~40μM时可以诱导p53的激活。
此后,该研究组[21]公布了在此三联苯骨架的基础上设计的一系列Bak-BH3 α-螺旋模拟物,其中显示出较好的体外活性的化合物的Ki值为0.114μM。
这些结果都可以说明以三联苯为骨架的α-螺旋模拟物对靶蛋白的抑制是有效的。
2.1.2 苯烯胺酮类Rodriguez等[22]通过把三联苯分子中间的苯环部分换成了烯胺酮结构,而烯胺酮的氨基能够与羰基形成分子内氢键,使得化合物的结构与苯环一样稳定。
2.1.3 联苯N, N-二甲基甲酰胺Rodriguez等[23]设计合成了一类二联苯4,4'-二甲基酰胺化合物来模拟α-螺旋的i,i+4,i+7,i+11位残基,具有更高的特异性和更好的选择性。
2.1.4 三联吡啶类较之苯环,杂环作为模拟α-螺旋的骨架小分子,会使得小分子的成药性更高,因为杂环的引入,连入疏水侧链的一面的对立面的亲水性比苯环更高,这增加了小分子整体的亲水性,使其在溶液中更加稳定。
前文提及,可以通过小分子三联苯化合物来模拟α-螺旋的骨架,Davis[24]为了拓宽这种α-螺旋骨架模拟的思路,想到可以用芳香基衍生物 (比如吡啶) 作为骨架,另外,在文献中也能查到一些关于二吡啶合成的方法,而其他取代模式的合成方法很少被提起。在此前提下,尝试合成了三联吡啶类化合物。
2.1.5 三吡啶酰胺类Ernst等[25]在总结了以α-螺旋来模拟小分子抑制剂的关键点 (能提供和母体蛋白相似角度关键氨基酸残基的模拟,易于合成,水溶性好) 后,尝试用酰胺类化合物来模拟α-螺旋。在此基础上,设计合成了以三吡啶聚酰胺为骨架的小分子α-螺旋模拟物。利用该模拟策略来设计Bak BH3/Bcl-XL抑制剂,并设计合成了一系列的衍生物,极化荧光结果显示,有三个以三吡啶酰胺为骨架的化合物显示出毫摩尔级的亲和性。
2.1.6 阶梯状多环醚类2005年,Oguri等[26]在观察阶梯状环醚海洋毒素的化学结构时发现它的拓扑结构和多肽的α-螺旋存在相似性,之后,为了能模拟这种类似的排列,通过SmI2介导的一系列醚类化合物的合成反应,合成了阶梯状多环醚类化合物,来模拟α-螺旋多肽。
2.1.7 咪唑类通过小分子来模拟α-螺旋时,考虑到三联苯类化合物较长,需要多步反应才能合成,存在得率低的缺点,德国的Antuch等[27]通过多组分反应合成了三取代的咪唑骨架,可以在两个取代基上连接苯环衍生侧链就可获得类似三联苯化合物,而合成该骨架只需要简单的一步反应即可完成。此外,这种具有广泛选择性的化合物前体对所合成的化合物的生物学和药代动力学活性都会有所改善。
2.2 模拟hotspots位于α-螺旋两面的小分子骨架 2.2.1 二联苯类在三联苯的基础上,Jacoby[28]总结了用小分子来模拟α-螺旋时,小分子应该具备的几个主要特征,提出可以用二联苯化合物模型来模拟α-螺旋中i,i+1,i+3和i+4四个氨基酸侧链的空间位置,该研究组建立了2, 6, 3′, 5′-二联苯取代模型来模拟α-螺旋。
同样,在提出的二联苯模型的基础上,Williams等[29]设计了一类二联苯雌激素受体共激活因子结合抑制剂 (CBIs)。通过汇聚式合成 (convergent) 方法,利用Suzuki偶联化学 (Suzuki coupling chemistry) 将各个模块单元相连接,实验结果也证实该类化合物能够竞争性结合雌激素受体共激活因子,但是亲和性和特异性还需要进一步改善。
2.2.2 联苯茚类Hamilton等[30]设计合成了以4, 7-二苯-1, 6-二取代茚为骨架的小分子来模拟α-螺旋上i,i+3,i+4和i+7残基。
2.3 模拟hotspots位于α-螺旋三面的小分子骨架三联苯类化合物的分子存在合成步骤长、不易更换取代基等缺点,而且,此类分子还只能模拟螺旋的两个疏水面,缺少对螺旋亲水一面hotspots的模拟,导致普遍存在亲和力低和特异性不足的缺陷, 面对这种局面,需要考虑的是增加小分子化合物对α-螺旋的第三面模拟。
2012年,Zhang等[31]在模拟Bim BH3α-螺旋来设计小分子抑制剂时发现,Bim BH3的α-螺旋hotspots分布在三个不同的面上。为了实现三面模拟,设计出一个在纳摩尔级的Bcl-2/Mcl-1小分子双重抑制剂,也提出在模拟α-螺旋设计小分子抑制剂时利用一个刚性的两元环结构来维持其三面结构稳定的骨架理念,在此理念的基础上,又设计出一个新型的两亲性三面模拟的小分子抑制剂,其对Mcl-1和Bcl-2的解离常数Ki分别为123nM和179nM[32]。2014年,该研究组的Li等[33]根据经验总结设计出一种十字型小分子骨架来模拟α-螺旋的三面和两圈构象,对Mcl-1和Bcl-2的解离常数分别为79nM和56nM。
Lee等[34]也考虑到了同样的问题,提出了另一种三面模拟α-螺旋设计小分子抑制剂的方法,即通过在模拟α-螺旋相反的一面介导引入合适的带电残基将小分子转化为两亲性的化合物。实验证明,相对于一面模拟的小分子化合物来说,这种三面模拟方式明显改善了小分子对靶标蛋白的亲和力,使其实现了对5个功能基团的模拟,即疏水面的i,i+3和i+7位氨基酸残基,亲水面的i-2和i+5位。而且,合成一个能够模拟α-螺旋三面的小分子化合物库对以后筛选高亲和力和高特异性的各种模型PPI抑制剂也是非常有益的。
在模拟设计的α-螺旋骨架的基础上,针对蛋白质-蛋白质相互作用的靶点,根据蛋白质表面的热点氨基酸残基分布,疏水空腔的深浅,在螺旋骨架的侧链连上合适的取代基,即可定向地设计抑制剂来干扰蛋白质-蛋白质的相互作用。所以,模拟α-螺旋多肽设计抑制剂也将成为一种新的抑制剂筛选策略。
3 结语通过多肽类和小分子类化合物来模拟α-螺旋设计PPI抑制剂在近几年发展迅速,通过模拟α-螺旋骨架上热点残基的方位分布及热点残基的空间结构,已经设计出多种可模拟不同方位热点残基的螺旋骨架和一系列对靶蛋白亲和力高的抑制剂,虽然到目前为止,还没有模拟α-螺旋设计的抑制剂进入临床阶段,但这两年设计合成了能够对α-螺旋骨架三面进行模拟的小分子化合物,为下一步利用这种骨架模型来建立更高选择性的化合物库奠定基础。这种通过模拟α-螺旋骨架来设计抑制剂的方法也为高亲和力和高特异性抑制剂的筛选提供了更多的选择性和可靠性,为抑制剂的设计提供了一种新的理念。
| [1] | Bullock B N, Jochim A L, Arora P S. Assessing helical protein interfaces for inhibitor design. Journal of the American Chemical Society, 2011, 133(36) : 14220–14223. DOI:10.1021/ja206074j |
| [2] | Kortemme T, Baker D. A simple physical model for binding energy hot spots in protein-protein complexes. Proceedings of the National Academy of Sciences, 2002, 99(22) : 14116–14121. DOI:10.1073/pnas.202485799 |
| [3] | Böttger V, Böttger A, Howard S F, et al. Identification of novel mdm2 binding peptides by phage display. Oncogene, 1996, 13(10) : 2141–2147. |
| [4] | Azzarito V, Long K, Murphy N S, et al. Inhibition of-helix-mediated protein-protein interactions using designed molecules. Nature Chemistry, 2013, 5(3) : 161–173. DOI:10.1038/nchem.1568 |
| [5] | 曹晨, 马堃. 蛋白质二级结构指定. 生物信息学, 2016, 14(3) : 181–187. Cao C, Ma K. Protein secondary structure assignment. Chinese Journal of Bioinformatics, 2016, 14(3) : 181–187. DOI:10.3969/j.issn.1672-5565.2016.03.09 |
| [6] | Wang D, Liao W, Arora P S, et al. Enhanced metabolic stability and protein-binding properties of artificial α helices derived from a hydrogen-bond surrogate:application to Bcl-xL. Angewandte Chemie, 2005, 44(40) : 6525–6529. DOI:10.1002/(ISSN)1521-3773 |
| [7] | Henchey L K, Porter J R, Ghosh I, et al. High specificity in protein recognition by hydrogen-bond-surrogate α-helices:selective inhibition of the p53/MDM2 complex. Chembiochem, 2010, 11(15) : 2104–2107. DOI:10.1002/cbic.v11:15 |
| [8] | Pellegrini M, Royo M, Chorev M, et al. Conformational consequences of i, i+3 cystine linkages:nucleation for α-helix. The Journal of Peptide Research, 1997, 49(5) : 404–414. |
| [9] | Shiau A K, Barstad D, Loria P M, et al. The structural basis of estrogen receptor/coactivator recognition and the antagonism of this interaction by tamoxifen. Cell, 1998, 95(7) : 927–937. DOI:10.1016/S0092-8674(00)81717-1 |
| [10] | Galande A K, Bramlett K S, Trent J O, et al. Potent inhibitors of LXXLL-based protein-protein interactions. ChemBioChem, 2005, 6(11) : 1991–1998. DOI:10.1002/cbic.v6:11 |
| [11] | Sia S K, Carr P A, Cochran A G, et al. Short constrained peptides that inhibit HIV-1 entry. Proceedings of the National Academy of Sciences, 2002, 99(23) : 14664–14669. DOI:10.1073/pnas.232566599 |
| [12] | Mauran L, Kauffmann B, Odaert B, et al. Stabilization of an α-helix by short adjacent accessory foldamers. Competes Rendus Chimie, 2016, 19(1-2) : 123–131. DOI:10.1016/j.crci.2015.07.003 |
| [13] | Tian Y, Wang D, Li J, et al. A proline-derived transannular N-cap for nucleation of short α-helical peptides. Chemical Communications, 2016, 52(59) : 9275–9278. DOI:10.1039/C6CC04672J |
| [14] | Wilson A J. Inhibition of protein-protein interactions using designed molecules. Chemical Society Reviews, 2009, 38(12) : 3289–3300. DOI:10.1039/b807197g |
| [15] | Cheng R P, Gellman S H, DeGrado W F. β-Peptides:from structure to function. Chemical Reviews, 2001, 101(10) : 3219–3232. DOI:10.1021/cr000045i |
| [16] | Lee E F, Sadowsky J D, Smith B J, et al. High-resolution structural characterization of a helical α/β-peptide foldamer bound to the anti-apoptotic protein bcl-xL. Angewandte Chemie, 2009, 121(24) : 4382–4386. DOI:10.1002/ange.v121:24 |
| [17] | Liu M, Li C, Pazgier M, et al. D-peptide inhibitors of the p53-MDM2 interaction for targeted molecular therapy of malignant neoplasms. Proceedings of the National Academy of Sciences, 2010, 107(32) : 14321–14326. DOI:10.1073/pnas.1008930107 |
| [18] | Grison C M, Miles J A, Robin S, et al. An α-helix-mimicking 12, 13-helix:selective protein-protein interaction inhibition using designed α/β/γ-foldamers. Angewandte Chemie, 2016, 55(37) : 11096–11100. DOI:10.1002/anie.201604517 |
| [19] | Orner B P, Ernst J T, Hamilton A D. Toward proteomimetics:terphenyl derivatives as structural and functional mimics of extended regions of an α-helix. Journal of the American Chemical Society, 2001, 123(22) : 5382–5383. DOI:10.1021/ja0025548 |
| [20] | Chen L, Yin H, Farooqi B, et al. p53α-Helix mimetics antagonize p53/MDM2 interaction and activate p53. Molecular Cancer Therapeutics, 2005, 4(6) : 1019–1025. DOI:10.1158/1535-7163.MCT-04-0342 |
| [21] | Yin H, Lee G, Sedey K A, et al. Terphenyl-based Bak BH3α-helical proteomimetics as low-molecular-weight antagonists of Bcl-xL. Journal of the American Chemical Society, 2005, 127(29) : 10191–10196. DOI:10.1021/ja050122x |
| [22] | Rodriguez J M, Hamilton A D. Intramolecular hydrogen bonding allows simple enaminones to structurally mimic the i, i+4, and i+7 residues of an α-helix. Tetrahedron Letters, 2006, 47(42) : 7443–7446. DOI:10.1016/j.tetlet.2006.08.048 |
| [23] | Rodriguez J M, Nevola L, Ross N T, et al. Synthetic inhibitors of extended helix-protein interactions based on a biphenyl 4, 4'-dicarboxamide scaffold. ChemBioChem, 2009, 10(5) : 829–833. DOI:10.1002/cbic.v10:5 |
| [24] | Davis J M, Truong A, Hamilton A D. Synthesis of a 2, 3'; 6', 3″-Terpyridine scaffold as an α-helix mimetic. Organic Letters, 2005, 7(24) : 5405–5408. DOI:10.1021/ol0521228 |
| [25] | Ernst J T, Becerril J, Park H S, et al. Design and application of an α-helix-mimetic scaffold based on an oligoamide-foldamer strategy:antagonism of the bak BH3/Bcl-xL complex. Angewandte Chemie, 2003, 115(5) : 553–557. DOI:10.1002/ange.200390122 |
| [26] | Oguri H, Oomura A, Tanabe S, et al. Design and synthesis of a trans-fused polycyclic ether skeleton as an α-helix mimetic scaffold. Tetrahedron Letters, 2005, 46(13) : 2179–2183. DOI:10.1016/j.tetlet.2005.02.039 |
| [27] | Antuch W, Menon S, Chen Q Z, et al. Design and modular parallel synthesis of a MCR derived α-helix mimetic protein-protein interaction inhibitor scaffold. Bioorganic & Medicinal Chemistry Letters, 2006, 16(6) : 1740–1743. |
| [28] | Jacoby E. Biphenyls as potential mimetics of protein α-helix. Bioorganic & Medicinal Chemistry Letters, 2002, 12(6) : 891–893. |
| [29] | Williams A B, Weiser P T, Hanson R N, et al. Synthesis of biphenyl proteomimetics as estrogen receptor-α coactivator binding inhibitors. Organic Letters, 2009, 11(23) : 5370–5373. DOI:10.1021/ol901999f |
| [30] | Hamilton A D, Kim I C. Diphenylindane-based proteomimetics reproduce the projection of the i, i+3, i+4, and i+7 residues on an α-helix. Organic Letters, 2006, 8(9) : 1751–1754. DOI:10.1021/ol052956q |
| [31] | Zhang Z, Li X, Song T, et al. An anthraquinone scaffold for putative, two-face Bim BH3α-helix mimic. Journal of Medicinal Chemistry, 2012, 55(23) : 10735–10741. DOI:10.1021/jm301504b |
| [32] | Zhang Z, Liang X, Li X, et al. Design and application of a rigid quinazolone scaffold based on two-face Bim α-helix mimicking. European Journal of Medicinal Chemistry, 2013, 69 : 711–718. DOI:10.1016/j.ejmech.2013.09.030 |
| [33] | Li X, Wang Z, Feng Y, et al. Two-face, two-turn α-helix mimetics based on a cross-acridine scaffold:analogues of the bim BH3 domain. ChemBioChem, 2014, 15(9) : 1280–1285. DOI:10.1002/cbic.v15.9 |
| [34] | Lee J H, Oh M, Kim H S, et al. Converting one-face α-helix mimetics into amphiphilic α-helix mimetics as potent Inhibitors of protein-protein interactions. ACS Combinatorial Science, 2015, 18(1) : 36–42. |