 2016, Vol. 36
2016, Vol. 36文章信息
- 万春红, 张志, 李圣纳, 彭以元, 许亮国
- WAN Chun-hong, ZHANG Zhi, LI Sheng-na, PENG Yi-yuan, XU Liang-guo
- TRAF7的研究进展
- Research Progresses on TRAF7
- 中国生物工程杂志, 2016, 36(3): 93-101
- China Biotechnology, 2016, 36(3): 93-101
- http://dx.doi.org/DOI:10.13523/j.cb.20160314
-
文章历史
- 收稿日期: 2015-10-08
- 修回日期: 2015-10-29






2. 江西师范大学功能有机小分子教育部重点实验室 南昌 330022
2. Key Laboratory of Fuctional Small Organic Molecule, Ministry of Education, Jiangxi Normal University, Nanchang 330022, China
肿瘤坏死因子受体相关因子(tumor necrosis factor receptor associated factor,TRAF)是机体抗病毒信号通路中重要的接头分子。在哺乳动物中,TRAFs通过结合TNF受体、TLRs/IL-1R(Toll-like/IL-l receptor)等多种受体家族转导上游信号,激活NF-κB(nuclear factor κB)、MAPK(mitogen-activated protein kinase)等不同的下游信号通路,从而参与调控免疫反应、细胞凋亡、应激反应和骨代谢等重要生理过程。目前,在哺乳动物中已发现该蛋白质在进化上非常保守,除了TRAF1的N端只有一个简单的锌指结构(zinc finger domain)外,其他TRAF分子的N端都含有一个RING指基序(RING-finger motif) 和若干个锌指结构域,其中TRAF2、TRAF3、TRAF6和TRAF7的RING指基序具有E3泛素连接酶活性,参与TRAF自身及靶蛋白的修饰。除TRAF7外,其他TRAFs家族成员都含有一个高度保守的C端CC结构域(coiled-coil domain),即TRAF结构域。该结构域又分成TRAF-N和TRAF-C,分别参与蛋白寡聚化,以及与受体家族和胞质因子的相互作用[1, 2, 3, 4]。
TRAF7最先是在TNFα诱导的NF-κB信号通路中筛选到的一种MEKK3的相互作用新蛋白[5]。而后通过生物信息学方法进一步鉴定出该蛋白质含有TRAF家族的RING结构域,将其命名为TRAF7,并发现其存在2.6kb和4.0kb两种形式的转录本,广泛表达于所有被检测组织中,其中在骨骼肌、心脏、胎盘等中的表达量高于在脑、胸腺、小肠和外周血淋巴细胞中的表达量[4]。此外,该蛋白质在一些疾病相关细胞中也有表达,并参与机体抗病原体感染信号通路的调节[6, 7]。由于其在信号通路中的重要作用,TRAF7的研究已引起了广泛关注,以下就目前的研究成果相对系统地介绍TRAF7的结构、功能及其互作蛋白,并分析相关的生物学过程。
1 TRAF7的结构TRAF7是TRAF蛋白家族的新成员,有两种剪接体,长的含670个氨基酸,称为TRAF7,短的相对少了N端66个氨基酸,称为TRAF7s。由图 1可见TRAF7在N端125~160氨基酸位点处有一个RING结构域,相邻221~287氨基酸处为锌指结构,通过基因库数据比对发现这两个结构域与TRAF家族高度保守,因而被归为TRAF家族[4, 5]。与TRAF2、TRAF3和TRAF6一样,TRAF7的RING结构域也含有E3泛素连接酶活性,为激活下游MAP3K家族激酶(MEKK1-3、NIK、ASK1和TAK1)所需[8, 9]。不同于其它TRAF家族成员,TRAF7的C端缺乏保守的TRAF结构域,却有1个CC结构域和7个WD40重复片段(外显子13~20),该片段与一些转导类蛋白Het-e、Het-d、F-box,以及一些含WD40结构的蛋白质FBXW6、FBXW7和FBX30高度同源。TRAF7不含传统的TRAF结构域,因此它的二聚化及胞中定位所需的结构位于一个含非经典的TRAF-N和CC结构域的中心区域[5]。
2 TRAF7的功能
TRAF7由于N端含有TRAF家族保守的RING和锌指结构域,所以也具有E3泛素连接酶活性。因其C端缺乏该家族保守的TRAF结构域,所以在信号转导过程中TRAF7不能直接结合模式识别受体,但是目前研究发现,TRAF7能以其独特机制转导TNFR和TLR受体介导的信号通路。以下从TRAF7的双重E3连接酶活性、其参与的信号通路及其相关的人类疾病三个方面进行阐述。
2.1 TRAF7的双重E3连接酶活性 2.1.1 TRAF7的泛素E3连接酶活性(1)MEKK3。Bouwmeester等[5]在构建人类TNFα/NF-κB信号转导的结构和功能图谱过程中,通过TAP标记的MEKK3分子筛选出FLJ33305(TRAF7)。用抗原表位标记的TRAF7进行免疫共沉淀实验发现内源性MEKK3能与TRAF7相互作用。共表达TRAF7与野生型或激酶失活型的MEKK3,发现MEKK3磷酸化TRAF7。将野生型或激酶失活型的MEKK3、TRAF7与His标记的泛素分子共转染,结果只有野生型MEKK3(不是激酶失活型MEKK3)能诱导TRAF7泛素化,说明磷酸化可能引发TRAF7的自身泛素化,有趣的是,这种泛素化不使TRAF7水解,而是增强TRAF7的稳定性。除MEKK3外,TRAF7是否与MAP3K家族其他成员相互作用?MEKK3能使TRAF7磷酸化/自泛素化,那么TRAF7是否影响MEKK3的激酶活性?Xu等[4]将HA-TRAF7分别与FLAG-MEKK2、MEKK3和RIP转染293细胞,然后通过免疫共沉淀结合体外激酶反应,发现TRAF7只特异性的与MEKK3相互作用,并增强了MEKK3的自身磷酸化活性,所以MEKK3很可能先使TRAF7磷酸化/自泛素化,然后稳定的TRAF7反过来增强MEKK3自身磷酸化。另外,通过构建TRAF7的缺失突变体,瞬时转染和免疫共沉淀实验显示,TRAF7的WD40重复区域(aa293~670)足以与MEKK3结合[4],并且MEKK3磷酸化TRAF7的N端[5]。
(2)与NEMO相互作用调节NF-κB信号通路。Tsikitis等[10]在TRAF7参与的肌细胞分化调控过程的研究中,稳定表达Flag-TRAF7,结合免疫亲和及离子交换色谱等方法鉴定出NEMO(NF-κB-essential modulator,IKKγ)与TRAF7相互作用,并且Traf7缺失后,大部分的p65处于细胞质中,推测TRAF7在肌细胞生成过程中通过修饰NEMO和影响p65的核转移来调节NF-κB活性,抑制肌细胞分化,但是TRAF7与NEMO、p65之间的具体作用机制还不清楚。为此,Zotti等[11]再一次通过系统的酵母双杂交筛选、免疫共沉淀和免疫印迹方法得出在TNF α刺激的HEK293细胞中TRAF7与NEMO存在相互作用。再通过使用不同大小的TRAF7或NEMO缺失突变体,发现NEMO通过1~230残基结合TRAF7的卷曲螺旋结构,在含RING指结构域的TRAF7突变体中,NEMO能被泛素化。反之,不能泛素化,说明在细胞内,TRAF7先通过CC结构域结合NEMO,再通过RING结构域泛素化NEMO,两者缺一不可。同理得出TRAF7与NF-κB的p65亚基相互作用,虽然结合位点未知。用含单一赖氨酸残基的泛素突变体做实验,发现NEMO和p65与TRAF7之间都是Lys-29连接的泛素化作用。细胞分馏实验中,在TNFα刺激下,不管是细胞质还是细胞核,p65似乎都被TRAF7泛素化,再一次验证TRAF7在细胞质和细胞核中都有定位[6, 12],而且p65在细胞质中含量更多。相反,共表达shRNA-TRAF7时,p65在细胞核中分布增多。综上所述,在TNFα诱导的HEK293/HeLa细胞中,TRAF7通过促进NEMO和p65的Lys29连接的泛素化及溶酶体降解,延迟p65的入核转移,抑制NF-κB激活,促使细胞凋亡[11]。
2.1.2 TRAF7的SUMO E3连接酶活性与TRAF2、TRAF6不同,TRAF7的RING结构域具有双重E3连接酶活性。另一种SUMO (small ubiquitin-like modifier)E3连接酶活性,与泛素化不同,SUMO化通常改变靶蛋白的亚细胞定位、蛋白质-DNA和蛋白质-蛋白质之间的相互作用。Morita等[6]在原癌基因产物c-Myb-鼠胚胎文库的酵母双杂交及免疫共沉淀实验中筛选并鉴定出TRAF7蛋白。TRAF7通过WD40重复区结合c-Myb的DNA结合结构域(DBD),激活c-Myb在Lys-523和Lys-499位点的SUMOs化,与p65相似,TRAF7的SUMO化使c-Myb 停留在细胞质内,抑制c-Myb诱导的反式活化,在c-Myb蛋白参与的造血干细胞的细胞分裂与分化中起负调节作用。值得注意的是,实验中观察到TRAF7在核膜边缘累积,说明TRAF7可能聚集在核膜上通过蛋白质修饰调节转录因子等的核转移。在目前发现的4种双功能的E3连接酶中,TOPORS和UHRF2的SUMO化不依赖于RING 结构域,TRIM27的SUMO化依赖RING结构域,虽然已知TRAF7通过WD40重复区与c-Myb结合,但是否依赖RING结构域还未知。而且有研究发现,UHRF2能自身SUMO化[13]。TRAF7能自泛素化,但目前还没有研究说明其能否自SUMO化。
2.2 TRAF7参与的信号转导途径信号转导途径是一个由一系列功能上相关联的蛋白质协调将环境信号转变成细胞表型反应的过程。胞外配体刺激受体后,经过一系列的受体近端分子(TRADD、FADD、MyD88等)、接头蛋白、MAPK家族和IKK激酶诱导一个信号级联反应,通过调节转录因子的活性,特异性的调控相关功能基因的表达。目前研究证明,TRAF7作为一种重要的接头蛋白,参与了TNFR和TLR2介导的信号通路中NF-κB、AP1和CHOP等转录因子的激活,如图 2所示。
2.2.1 TNFR介导的信号通路
TRAF7主要参与TNFα配体刺激下TNFR1介导的信号通路。首先是NF-κB通路,Zotti 等[11]在NF-κB luci荧光报告实验中发现,不管是内源刺激(TNFα、IL-1β和LPS),还是外界转染NF-κB激活子,TRAF7表达都显著降低了NF-κB的活性。用慢病毒shRNA敲除TRAF7后,NF-κB的活性都增加,说明TRAF7通过泛素化NEMO和p65,参与并抑制了TNFR介导的NF-κB的激活。Tsikitis等[10]在肌细胞生成方面也有类似研究,但不同的是,Tsikitis等的实验发现TRAF7对NEMO和p65的泛素化却激活了NF-κB。是不同的细胞环境,还是不同的生理条件所致?还需要进一步研究。其次是AP1和CHOP通路,之前有研究表明MEKK3参与JNK和p38信号途径,分别激活AP1和CHOP[14],那么TRAF7作为MEKK3的互作蛋白质是否也参与其中?为此Xu等[4]通过AP1和CHOP的报告基因实验发现TRAF7自身不激活AP1或微弱激活CHOP,然而TRAF7却能增强MEKK3诱导的AP1和CHOP激活,并且还存在剂量效应。实验中,TRAF7在MEKK1介导的AP1和CHOP激活方面无显著影响。但奇怪的是,反义RNA-TRAF7没有影响MEKK1诱导的AP1激活,却抑制了MEKK1诱导的CHOP激活。Bouwmeester等[5]表明单独过表达TRAF7或缺失突变体不激活NF-κB,然而,共表达野生型TRAF7(突变体不行)和MEKK3导致了NF-κB(20倍)和AP1(3倍)的协同激活。大多数情况下,这种协同激活依赖MEKK3的激酶活性。这说明TRAF7参与的TNFR-NF-κB和AP1/CHOP是相互关联的,根据不同的细胞环境和生物背景,TRAF7可能对NF-κB有一个竞争性的激活或抑制作用。
需要补充的是,TNFα具有细胞毒性,其刺激后的生理效应取决于促生存因子(NF-κB)和促凋亡因子(JNK)之间的平衡。最初,Xu等[4]通过瞬时转染等实验发现,TRAF7通过RING结构域诱导caspase依赖的细胞凋亡。之后,Scudiero等[15]对TRAF7的促凋亡机制进行了详细研究,其中涉及一种重要的抗凋亡蛋白c-FLIP(CASP8 and FADD-like apoptosis regulator),它能通过干扰caspase在DISC上的激活来抑制细胞凋亡。研究中发现,TRAF7可通过三条途径降低c-FLIP的量,从而诱导细胞凋亡。其一,c-FLIP的表达受一些转录因子的调节,包括NF-κB和p53。TRAF7表达后,NF-κB受抑制,c-FLIP的表达下调。其二,在TNFα刺激下,TRAF7诱导激活JNK,JNK激活Itch,Itch泛素化c-FLIPL ,转而进入蛋白酶体降解[16]。其三,TRAF7自身与c-FLIPL 存在Lys-48/29/27连接的泛素化作用,可以通过Lys-48-linked泛素化,使c-FLIPL进入蛋白酶体降解,或通过Lys-29-linked泛素化,进入溶酶体降解。TRAF7通过这些使细胞对外界刺激(TNFα毒性)更加敏感,促进细胞凋亡。临床前数据显示,在恶性肿瘤中经常发现c-FLIPL的表达量增加[17],所以选择性的使用c-FLIP的抑制剂,结合TRAF7对c-FLIPL的调节,为抗肿瘤治疗提供了新的途径。
2.2.2 TLR2介导的信号通路TLRs是一类从线虫到哺乳动物都保守的模式识别受体,通过识别配体引发各种免疫反应和炎症反应以抵抗病原体侵染。Yoshida等[12]的研究发现,TRAF7/TRAF6主要参与TLR2-NF-κB信号途径,对AP1或其他转录因子很少报道,而且单独过表达wtTRAF7不能增强NF-κB激活,共表达时可以协同增强TRAF6诱导的NF-κB激活,说明TRAF7可作为共转导子增强TRAF6诱导的TLR2-NF-κB激活。与此同时,TLRs通常与自身免疫、慢性炎症和感染性疾病的发病机制有关,因此其反馈调节十分重要。目前有两种去泛素化酶(DUBs)在IKK上游抑制NF-κB活性,其中一个就是圆柱瘤肿瘤抑制蛋白CYLD(cylindromatosis),CYLD的基因突变能引发家族性圆柱瘤的形成[18]。该研究发现,在TLR2配体刺激下,TRAF6或TRAF7泛素化增强,经过IKKs-IκBα和MKK3/6-p38激活NF-κB通路,进而诱导CYLD连同其他炎症介质(TNF-α、IL-1β、IL-8)的转录。CYLD反过来使TRAF6和TRAF7去泛素化,从而抑制NF-κB激活,在TLR2依赖的NF-κB信号通路中起自反馈调节作用[12]。另一项研究也有类似结果,CYLD使TRAF6/7去泛素化,抑制非分型流感嗜血杆菌(NTHi)诱导的TLR2-NF-κB通路,从而帮助宿主对抗NTHi感染导致的不良炎症反应。其实除了TLR2,用R837(一种TLR7的配体)刺激HEK293细胞,共表达TRAF7的负显性突变体后,NF-κB的激活也受到影响,说明TRAF7可能还参与了其他TLR家族成员的信号通路[7]。
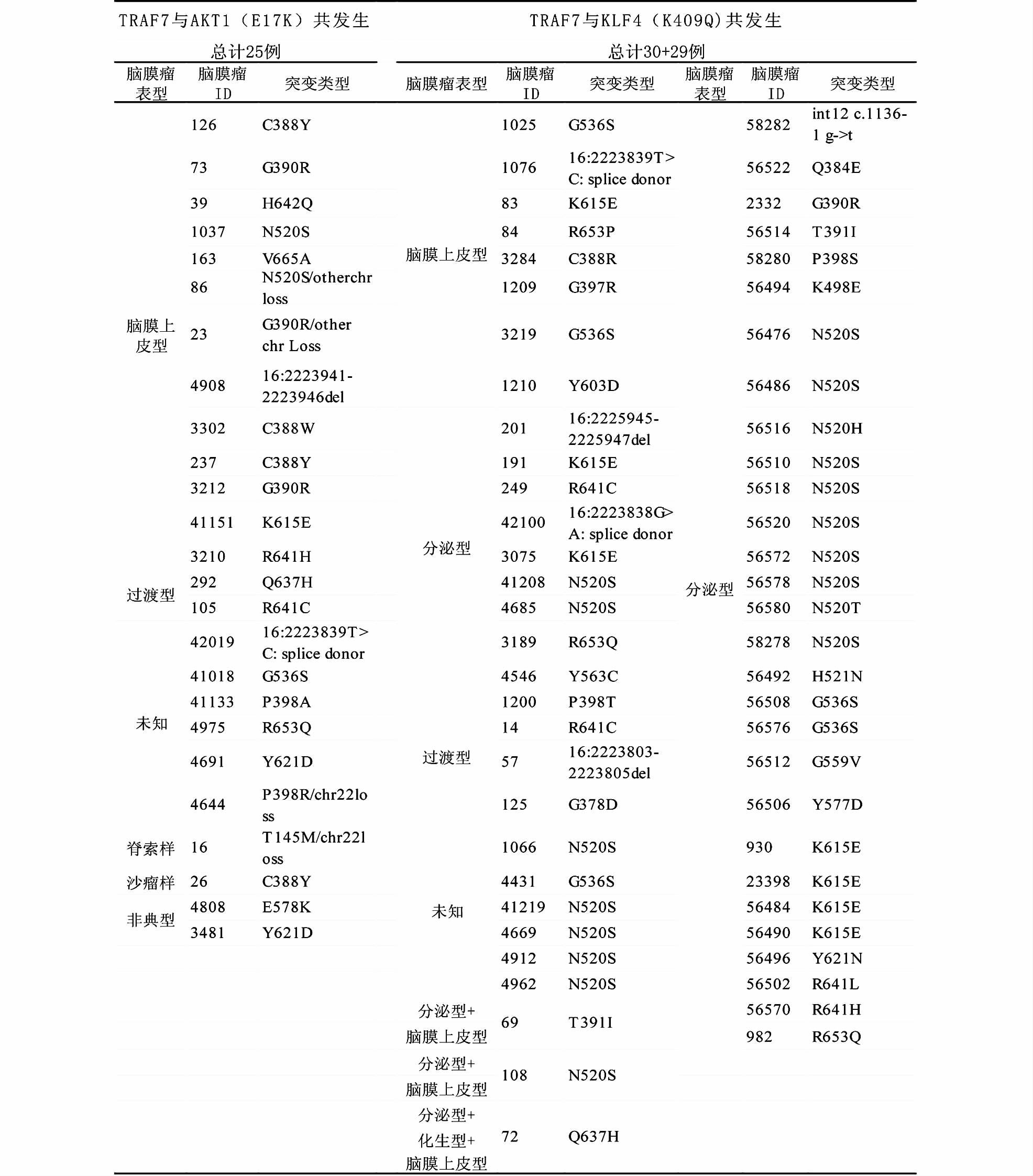
2.3 TRAF7相关的人类疾病 2.3.1 TRAF7与脑膜瘤(meningiomas)脑膜瘤与TRAF7的关系是近年研究最多的课题之一。脑膜瘤起源于中枢神经系统的脑膜区,是最常见的原发性脑肿瘤,大部分脑膜瘤患者存在NF2(neurofibromatosis type 2)基因的突变,该突变破坏了肿瘤抑制基因Merlin的正常表达,进而导致肿瘤的发生。根据临床观察、基因表达和H3K27乙酰化作用的不同,将脑膜瘤分为NF2/chr22loss型和非NF2突变型。为了全面描述脑膜瘤的基因组学特征和进一步了解肿瘤形成的分子机制,Clark等[19]先用50例非辐射的I级(n=39)和II级(n=11)脑膜瘤进行基因型分型和外显子测序,鉴定出除NF2基因突变外还存在其他几种基因的突变。他们从中选取前5个基因(NF2、TRAF7、KLF4、SMO和AKT1),再用另外250例非辐射的脑膜瘤(204例I级、44例II级、2例III级)进行基因定向重排,并分析22号染色体拷贝数。同时,Reuss等[20]用30例分泌型脑膜瘤和89例非分泌型脑膜瘤分析了TRAF7和KLF4在特定类型脑膜瘤中的分布及功能。
在以上300例脑膜瘤中,近50%是NF2基因失活型(149例染色体22丢失,其中包括108例NF2基因突变),在另外50%非NF2型脑膜瘤中,如表 1所示,AKT1(n=38,13%)突变类型为E17K,有28例与TRAF7突变共发生;SMO(n=11,4%)突变类型为L412F和W535L,只有1例与TRAF7共发生;KLF4(n=31,10%)突变类型为K409Q,有30例与TRAF7共发生;TRAF7(n=72,24%)的突变位点如图 3所示,除与其他突变共存外,有14例脑膜瘤中只有TRAF7突变,并且TRAF7、KLF4、AKT1突变基本上与NF2突变不能共存[19]。Reuss等[20]的实验显示,所有的分泌型脑膜瘤都含有KLF4(30/30)和TRAF7突变(29/30),89例非分泌型脑膜瘤中并未发现KLF4突变,5/21脑膜上皮型和2/20非典型脑膜瘤中发现了TRAF7突变,所以KLF4只存在于分泌型脑膜瘤中。但矛盾的是,如表 1所示,在Clark等[19]的研究结果中,脑膜上皮型等非分泌型脑膜瘤中仍有KLF4突变。TRAF7的突变基本是单个核苷酸的改变,但也有少数几个短序列的改变。除了SMO,大多数AKT1突变和近全部的KLF4突变都与TRAF7突变共发生,由图 3可知,TRAF7突变基本分布于WD40重复区域,而且分泌型脑膜瘤的突变主要位于TRAF7的第4个WD40区,在整个脑膜瘤生成中,N520也是突变频率最高的位点。


目前已发现脑膜瘤与多种信号通路相关,虽然具体机制还需进一步研究。例如,脑膜瘤细胞通过RAS/MAPK和PI3K/AKT信号途径增强一些生长因子(如PDGF[21]、IGF[22])的表达,激活自分泌环,诱导肿瘤细胞生长、迁移和血管生成。在细胞分裂过程中,RB抑制转录因子E2F,阻断细胞周期由G1期向S期转变,当RB功能异常时,活化的E2F磷酸化p14,活化的p14抑制p53的MDM2降解,p53进一步通过p21抑制细胞周期进行,促进细胞凋亡。但在脑膜瘤细胞中,由于9号染色体上的三个抑癌基因(CDKN2A/B/C)失活突变,导致RB和p53异常,细胞无限增殖[23]。此外,脑膜瘤的发生还与其他信号通路有关。例如,由EGF、VEGF生长因子诱导的PLCγ1/IP3/DAG通路和PLA2/COX2通路,与SMO突变有关的Hh信号通路[24]。虽然这些信号途径基本上都是通过促进细胞生长、增殖、迁移,抑制细胞凋亡,有的还促进血管生成,或影响基质重塑和干细胞稳态(Hh途径)等来导致脑膜瘤发生,但对于TRAF7具体的参与机制,目前还没有研究。根据TRAF7能与MEKK3、p53互作,并且与AKT1、SMO突变共发生,我们可以推测在脑膜瘤细胞中,TRAF7突变可能参与了RAS/MAPK信号途径、p53的功能异常及Hh通路。当然,TRAF7也可以直接发挥作用。例如,TRAF7突变后,caspase依赖的细胞凋亡受抑制,细胞得以继续增殖。
这些基因的突变与组织亚型也存在一定关系,NF2/chr22lose型脑膜瘤主要分布于大脑半球,而TRAF7等4种基因突变相关的脑膜瘤主要分布于颅底的中间区[19]。根据这些基因突变的特异性的分子结构、组织分布、表型特征等,我们可以预测突变概况及可能的药物反应,再通过建立动物模型进行临床前药物测试,及早实现药物靶向治疗。
2.3.2 TRAF7与乳腺癌在正常细胞内,肿瘤抑制因子p53主要通过诱导Mdm2表达,Mdm2作为E3连接酶泛素化修饰p53,抑制p53的活性,使p53低水平存在,细胞正常增殖。在应激条件下,如DNA损伤时,p53蛋白磷酸化,滞留于细胞核内,不与Mdm2结合,在细胞中含量增加,造成细胞周期停顿,启动DNA修复有关的基因表达。如果DNA损伤程度过高,p53直接诱导细胞凋亡[25]。由此可知,p53的基本功能是捍卫基因组的完整性,在细胞凋亡、DNA修复和细胞周期中发挥它的抗癌作用,因此它的功能异常与多种癌症有关。
早在对乳腺癌的研究中就发现了p53的突变,该突变破坏了p53的MDM2降解[26]。最近在有些乳腺癌样本中,发现p53蛋白在细胞质中大量积累,却没有发现p53的突变,说明还存在其他的途径使p53的胞质含量上升。故用qRT-PCR检测E3连接酶的mRNA的表达,结果发现,TRAF7 mRNA的量在乳腺肿瘤中比相邻组织中大大降低了。进一步体外泛素化实验表明,在TRAF7存在下,p53发生了K48连接的多聚泛素化。相反,当转染缺乏RING结构域的TRAF7突变体时,p53未泛素化,说明TRAF7通过RING结构域泛素化p53,并使p53降解。因此可以得出在乳腺癌中,TRAF7含量下降,阻碍了对p53的泛素化降解,导致p53在细胞质中累积,影响了其正常的转录因子活性,共同导致了癌症的发生。因此,TRAF7含量下降和p53细胞质累积都可以作为乳腺癌风险预测的指标。例如,通过Kaplan-Meier生存曲线发现,TRAF7lowp53high的存活率低于TRAF7highp53low的[27]。有研究发现在乳腺癌细胞中,另一种双功能的E3连接酶UHRF2也能间接或直接调节一些关键因子(如p53和pRb)的功能[28]。
综合p53的Mdm2和TRAF7泛素化修饰过程,可以得出p53的泛素蛋白酶体降解途径与其转录反应具有功能性联系,具体生化机制还不清楚。作为研究最多的肿瘤之一,乳腺癌的发生与HER、Notch、Wnt等信号通路相关[29]。那么在乳腺癌的发生过程中,TRAF7除了影响p53的降解外,是否还参与其他信号通路?是什么因素导致了乳腺癌中TRAF7的表达下调,目前还没有明确解释。
2.3.3 TRAF7与其他疾病作为参与调节先天免疫及适应性免疫的重要分子,TRAF7与多种疾病的发生有关,其中包括一些目前研究不多的疾病。例如,“TSC2-PKD1连续缺失综合征”,其中结节性硬化症[30]和双侧多囊性肾病(ADPKD)都是人类常见的常染色体遗传病。“TSC2-PKD1连续缺失综合征”是指同时缺失了TSC2基因和PKD1基因,这一类患者表现出两者的综合症状。Boehm等[31]通过断点分析和qPCR 等方法发现综合征患者的染色体上除了缺失整个TSC2和PKD1以外,下游RAB26、TRAF7和大部分CASKIN1基因都被删除了,说明TRAF7与“TSC2-PKD1连续缺失综合征”具有某种关联,只是相关研究还不多,具体机制仍不清楚。之前研究提出在肌细胞中,肌原性调节因子MyoD1(Myoblast determination protein 1)能够结合TRAF7的启动子,在转录水平调节TRAF7的表达[10]。最近有研究发现,在静脉内皮细胞中,miR-126直接作用于TRAF7 mRNA上的3′UTR(GGUACGAC),在转录后水平抑制TRAF7的表达。内皮细胞的凋亡是动脉粥样硬化、斑块破裂等病发的重要因素[32],miR126通过降低TRAF7表达和ROS形成来抑制细胞凋亡,这可能成为预防和治疗动脉粥样硬化等血管疾病的潜在目标[33]。
3 小 结TRAF蛋白家族是一类在哺乳动物中发现的遗传学上保守的胞内接头蛋白,通过和受体结合传递上游信号激活NF-κB、AP-1等转录因子,调控相关基因的表达,从而影响细胞的生存、增殖、分化、死亡和多个生物学的调控过程。TRAF7自鉴定以来,已基本掌握其结构特点,但是功能研究还不完善,目前仅发现TRAF7参与TNFR1和TLR2两条信号通路,且机制认识还不具体。例如,TRAF7缺乏保守的TRAF结构域,不能直接结合受体,其可能的替代途径是什么?虽然不少研究证实TRAF7与固有免疫、肿瘤发生等多种生命活动现象有关,但是对其中涉及的TRAF7功能和机制仍存在许多疑问,TRAF7基因在脑膜瘤和TSC2-PKD1综合征中突变后的具体影响是什么?是否改变了细胞信号途径或相关分子表达模式?对于这些还需对TRAF7做更进一步的研究,不断完善TRAF7的细胞功能图谱,早日为相关疾病的预防和治疗提供理论依据。
致谢 感谢江西师范大学博士启动基金对本研究的资助。
| [1] | Bradley J R, Pober J S. Tumor necrosis factor receptor-associated factors (TRAFs). Oncogene, 2001, 20(44):6482-6491. |
| [2] | Chung J Y, Park Y C, Ye H, et al. All TRAFs are not created equal: common and distinct molecular mechanisms of TRAF-mediated signal transduction. J Cell Sci, 2002, 115(Pt 4):679-688. |
| [3] | Thomas G S, Zhang L, Blackwell K, et al. Phosphorylation of TRAF2 within its RING domain inhibits stress-induced cell death by promoting IKK and suppressing JNK activation. Cancer Res, 2009, 69(8):3665-3672. |
| [4] | Xu L G, Li L Y, Shu H B. TRAF7 potentiates MEKK3-induced AP1 and CHOP activation and induces apoptosis. J Biol Chem, 2004, 279(17):17278-17282. |
| [5] | Bouwmeester T, Bauch A, Ruffner H, et al. A physical and functional map of the human TNF-alpha/NF-kappa B signal transduction pathway. Nat Cell Biol, 2004, 6(2):97-105. |
| [6] | Morita Y, Kanei-Ishii C, Nomura T, et al. TRAF7 sequesters c-Myb to the cytoplasm by stimulating its sumoylation. Mol Biol Cell, 2005, 16(11):5433-5444. |
| [7] | Zimmer J, Lim J H, Jono H, et al. Tumor suppressor CYLD acts as a negative regulator for non-typeable haemophilus influenza-induced inflammation in the middle ear and lung of mice. PLoS One, 2007, 2(10):e1032. |
| [8] | Alvarez S E, Harikumar K B, Hait N C, et al. Sphingosine-1-phosphate is a missing cofactor for the E3 ubiquitin ligase TRAF2. Nature, 2010, 465(7301):1084-1088. |
| [9] | Kayagaki N, Phung Q, Chan S, et al. DUBA: a deubiquitinase that regulates type I interferon production. Science, 2007, 318(5856):1628-1632. |
| [10] | Tsikitis1 M, Acosta-Alvear D, Blais A, et al. Traf7, aMyoD1 transcriptional target, regulates nuclear factor-kB activity duringmyogenesis. EMBO Mol Med, 2010, 11(12):969-976. |
| [11] | Zotti T, Uva A, Ferravante A, et al. TRAF7 protein promotes Lys-29-linked polyubiquitination of IkappaB kinase (IKKgamma)/NF-kappaB essential modulator (NEMO) and p65/RelA protein and represses NF-kappaB activation. J Biol Chem, 2011, 286(26):22924-22933. |
| [12] | Yoshida H, Jono H, Kai H, et al. The tumor suppressor cylindromatosis (CYLD) acts as a negative regulator for toll-like receptor 2 signaling via negative cross-talk with TRAF6 AND TRAF7. J Biol Chem, 2005, 280(49):41111-41121. |
| [13] | Oh Y, Chung K C. UHRF2, a ubiquitin E3 ligase, acts as a small ubiquitin-like modifier E3 ligase for zinc finger protein 131. J Biol Chem, 2013, 288(13):9102-9111. |
| [14] | Nakamura K, Johnson G L. PB1 domains of MEKK2 and MEKK3 interact with the MEK5 PB1 domain for activation of the ERK5 pathway. J Biol Chem, 2003, 278(39):36989-36992. |
| [15] | Scudiero I, Zotti T, Ferravante A, et al. Tumor necrosis factor (TNF) receptor-associated factor 7 is required for TNFalpha-induced Jun NH2-terminal kinase activation and promotes cell death by regulating polyubiquitination and lysosomal degradation of c-FLIP protein. J Biol Chem, 2012, 287(8):6053-6061. |
| [16] | Chang L, Kamata H, Solinas G, et al. The E3 ubiquitin ligase itch couples JNK activation to TNFalpha-induced cell death by inducing c-FLIP(L) turnover. Cell, 2006, 124(3):601-613. |
| [17] | Yang J K. FLIP as an anti-cancer therapeutic target. Yonsei Medical Journal, 2008, 49(1):19. |
| [18] | Chen Z J. Ubiquitin Signaling in the NF-κB Pathway. Nat Cell Biol, 2005, 7(8):758-765. |
| [19] | Clark V E, Erson-Omay E Z, Serin A, et al. Genomic analysis of non-NF2 meningiomas reveals mutations in TRAF7, KLF4, AKT1, and SMO. Science, 2013, 339(6123):1077-1080. |
| [20] | Reuss D E, Piro R M, Jones D T, et al. Secretory meningiomas are defined by combined KLF4 K409Q and TRAF7 mutations. Acta Neuropathol, 2013, 125(3):351-358. |
| [21] | Johnson M D, O'Connell M J, Pilcher W, et al. Fibroblast growth factor receptor-3 expression in meningiomas with stimulation of proliferation by the phosphoinositide 3 kinase-Akt pathway. J Neurosurg, 2010, 112(5):934-939. |
| [22] | Wrobel G, Roerig P, Kokocinski F, et al. Microarray-based gene expression profiling of benign, atypical and anaplastic meningiomas identifies novel genes associated with meningioma progression. Int J Cancer, 2005, 114(2):249-256. |
| [23] | Goutagny S, Yang H W, Zucman-Rossi J, et al. Genomic profiling reveals alternative genetic pathways of meningioma malignant progression dependent on the underlying NF2 status. Clin Cancer Res, 2010, 16(16):4155-4164. |
| [24] | Brastianos P K, Horowitz P M, Santagata S, et al. Genomic sequencing of meningiomas identifies oncogenic SMO and AKT1 mutations. Nat Genet, 2013, 45(3):285-289. |
| [25] | Kim E S, Shohet J M. Reactivation of p53 via MDM2 inhibition. Cell Death Dis, 2015, 6(10):e1936. |
| [26] | Patocs A, Zhang L, Xu Y, et al. Breast-cancer stromal cells with TP53 mutations and nodal metastases. N Engl J Med, 2007, 357(25):2543-2551. |
| [27] | Wang L, Wang L, Zhang S, et al. Downregulation of ubiquitin E3 ligase TNF receptor-associated factor 7 leads to stabilization of p53 in breast cancer. Oncol Rep, 2013, 29(1):283-287. |
| [28] | Wu J, Liu S, Liu G, et al. Identification and functional analysis of 9p24 amplified genes in human breast cancer. Oncogene, 2012, 31(3):333-341. |
| [29] | Nwabo Kamdje A H, Seke Etet P F, Vecchio L, et al. Signaling pathways in breast cancer: therapeutic targeting of the microenvironment. Cell Signal, 2014, 26(12):2843-2856. |
| [30] | Napetschnig J, Wu H. Molecular basis of NF-kappaB signaling. Annu Rev Biophys, 2013, 42:443-468. |
| [31] | Boehm D, Bacher J, Neumann H P H. Gross genomic rearrangement involving the TSC2-PKD1 contiguous deletion syndrome: characterization of the deletion event by quantitative polymerase Chain reaction deletion assay. American Journal of Kidney Diseases, 2007, 49(1):e11-e21. |
| [32] | Libby P, Ridker P M, Hansson G K. Progress and challenges in translating the biology of atherosclerosis. Nature, 2011, 473(7347):317-325. |
| [33] | Wang Y, Wang F, Wu Y, et al. MicroRNA-126 attenuates palmitate-induced apoptosis by targeting TRAF7 in HUVECs. Mol Cell Biochem, 2015, 399(1-2):123-130. |